Distinguishing Physisorbed and Chemisorbed Species with XPS: A Guide for Biomaterial and Drug Development Research
This article provides a comprehensive guide for researchers on using X-ray Photoelectron Spectroscopy (XPS) to analyze surface-adsorbed species.
Distinguishing Physisorbed and Chemisorbed Species with XPS: A Guide for Biomaterial and Drug Development Research
Abstract
This article provides a comprehensive guide for researchers on using X-ray Photoelectron Spectroscopy (XPS) to analyze surface-adsorbed species. It begins by establishing the critical differences between physisorption and chemisorption, explaining their distinct spectral signatures in XPS data. It then details practical methodologies for sample preparation, data acquisition, and peak fitting specific to adsorbed layers. The guide addresses common challenges in data interpretation, offering troubleshooting and optimization strategies to enhance signal clarity and avoid contamination artifacts. Finally, it explores validation techniques and comparative analyses, highlighting the role of complementary surface science methods. Tailored for biomedical and pharmaceutical scientists, this resource empowers accurate surface characterization of drug delivery systems, implants, and diagnostic platforms.
Understanding Adsorption on Surfaces: Physisorption vs. Chemisorption and Their XPS Signatures
In a thesis investigating surface species via X-ray Photoelectron Spectroscopy (XPS), distinguishing between physisorption and chemisorption is paramount. This differentiation dictates sample preparation, ultra-high vacuum (UHV) compatibility, data interpretation, and the understanding of surface reactivity. Physisorbed species, bound by weak van der Waals forces, are often considered contaminants in XPS, while chemisorbed species, forming chemical bonds, are the primary subject of surface chemistry studies.
Comparative Analysis
Table 1: Fundamental Characteristics of Physisorption vs. Chemisorption
| Parameter | Physisorption | Chemisorption |
|---|---|---|
| Binding Energy | Very low (< 50 kJ/mol) | High (40-800 kJ/mol) |
| Specificity | Non-specific | Highly specific to surface sites/chemistry |
| Temperature Range | Occurs at/below condens. temp. of adsorbate | Can occur at higher temperatures |
| Reversibility | Fully reversible | Often irreversible or requires high energy for desorption |
| Layer Thickness | Multilayers possible | Typically limited to a monolayer |
| XPS Spectral Impact | Can cause charging, mask signals; often removed by UHV/mild heating. | Causes identifiable chemical shifts in core-level peaks. |
| Adsorption Isotherm | Fits Langmuir or BET models for multilayers | Typically fits Langmuir isotherm (monolayer) |
Table 2: XPS-Specific Observations and Protocols
| Aspect | Physisorbed Species | Chemisorbed Species |
|---|---|---|
| Sample Handling | Requires cryo-transfer or in-situ dosing to study. | Standard UHV-compatible preparation. |
| In-Situ Cleaning | Removed by gentle heating (often < 150°C) or prolonged UHV. | Not removed by mild heating; may decompose or react. |
| Spectral Evidence | Little to no change in substrate peak BE; may add adventitious C 1s, O 1s. | Observable chemical shift in substrate/adsorbate peaks; new bonding states. |
| Quantification | Difficult, often inconsistent due to desorption. | Reliable, used for coverage calculation. |
Experimental Protocols for XPS-Based Research
Protocol 1: Differentiating Physisorbed vs. Chemisorbed Water on Metal Oxides
Objective: To identify the nature of adsorbed water/hydroxyl species on TiO₂ using XPS and temperature-programmed desorption (TPD) within the XPS chamber.
- Sample Preparation: Clean TiO₂ single crystal or thin film by cycles of Ar⁺ sputtering (1 keV, 5 µA, 10 min) and annealing at 600°C in UHV (base pressure < 5x10⁻¹⁰ mbar) to obtain a clean, ordered surface.
- Dosing: Expose the clean surface to deuterated water (D₂O) at 100 K using a directed doser. Dose is quantified in Langmuirs (1 L = 10⁻⁶ Torr·sec).
- XPS Analysis (100 K): Acquire high-resolution O 1s and Ti 2p spectra.
- Expected: A broad O 1s peak with components at ~533.0 eV (physisorbed D₂O) and ~531.5 eV (chemisorbed OD).
- Temperature-Programmed XPS:
- Warm the sample in stages (100 K → 150 K → 300 K → 500 K) using a resistive heater.
- At each stage, acquire O 1s spectra.
- Expected: The ~533.0 eV component diminishes by 150 K (physisorption). The ~531.5 eV component remains until >300 K (chemisorption).
- Data Analysis: Plot the integrated intensity of each O 1s component versus temperature to generate a desorption profile.
Protocol 2: Studying Chemisorption of Functional Groups on Drug Delivery Nanoparticles
Objective: To characterize the covalent grafting of silane-PEG ligands onto silica nanoparticles (SiNPs) for drug delivery.
- Functionalization: React purified SiNPs (50 nm) with 3-(aminopropyl)triethoxysilane (APTES, 2% v/v in anhydrous toluene) under reflux for 6 hours. Centrifuge and wash thoroughly with toluene and ethanol.
- XPS Sample Preparation: Drop-cast a concentrated dispersion of functionalized SiNPs onto an indium foil or a gold-coated substrate. Dry under inert atmosphere.
- Control Sample: Prepare a sample of unmodified SiNPs treated only with toluene.
- XPS Analysis:
- Acquire high-resolution spectra of Si 2p, O 1s, C 1s, and N 1s regions.
- For unmodified SiNPs: C 1s shows only adventitious hydrocarbon (C-C/C-H at 284.8 eV).
- For APTES-modified SiNPs: The N 1s peak at ~399.5 eV confirms chemisorbed amine. Deconvolution of C 1s shows new components for C-N (~286.0 eV) and C-O (from possible PEG, ~286.5 eV).
- Quantification: Calculate the atomic % of N. Estimate surface coverage using known nanoparticle surface area.
Visualization
Title: XPS Workflow for Differentiating Adsorption Types
Title: Energy Landscape Comparison of Adsorption Types
The Scientist's Toolkit: Research Reagent Solutions & Essential Materials
Table 3: Key Materials for Adsorption & XPS Studies
| Item | Function & Relevance |
|---|---|
| UHV-Compatible Sample Holder | Allows resistive heating and cooling (to 100K) of samples inside the XPS chamber for in-situ studies. |
| Directed Gas/Liquid Dosers | For controlled exposure of surfaces to vapors (e.g., H₂O, organics) or reactive gases (e.g., O₂, H₂) without contaminating the entire chamber. |
| Deuterated Water (D₂O) | Used in TPD/XPS studies to distinguish adsorbed water from background water vapor signals via mass spectrometry or isotopic shift. |
| Functional Silanes (e.g., APTES) | Standard reagents for covalent (chemisorbed) functionalization of oxide surfaces (SiO₂, TiO₂) to model drug carrier coatings. |
| Single Crystal Substrates (Au(111), TiO₂(110)) | Well-defined surfaces essential for fundamental adsorption studies, providing reproducible sites and minimal roughness. |
| Argon Gas (99.999%) | Source gas for ion sputtering guns used to clean sample surfaces in-situ prior to adsorption experiments. |
| Indium Foil | A ductile, conductive substrate for mounting powder samples like nanoparticles for XPS analysis. |
| Calibrated Leak Valves | Precisely control the introduction of research-grade gases into the UHV chamber for quantitative dosing. |
Why XPS is a Critical Tool for Surface Adsorption Analysis
X-ray Photoelectron Spectroscopy (XPS) is a cornerstone technique in surface science, enabling the quantitative and chemical-state analysis of the outermost atomic layers (1-10 nm) of a material. Within the broader thesis on XPS analysis for physisorbed and chemisorbed species research, its criticality stems from its unique ability to differentiate between adsorption modes. XPS provides direct evidence of chemical bond formation (chemisorption) via measurable shifts in binding energy, while also quantifying monolayer coverage and the elemental composition of physisorbed layers. This application note details protocols and data for utilizing XPS in adsorption studies relevant to catalysis, sensor development, and drug delivery systems.
Core Principles and Data Sensitivity
XPS probes adsorption by detecting changes in surface composition and electronic structure. Key quantitative outputs include:
- Binding Energy (BE) Shifts: > 0.3 eV shift typically indicates chemisorption and new chemical state formation.
- Coverage (θ) Calculation: Derived from attenuated substrate signal or increased adsorbate signal.
- Atomic Concentration (%): Direct measure of surface elemental composition post-adsorption.
Table 1: Characteristic XPS Signatures for Adsorption Types
| Adsorption Type | Bond Nature | Typical BE Shift of Substrate/Adsorbate | Key Spectral Evidence |
|---|---|---|---|
| Physisorption | Van der Waals, electrostatic | Minimal (< 0.2 eV) | Attenuation of substrate peaks; no new chemical states. |
| Chemisorption | Covalent/Ionic bond formation | Significant (> 0.3 eV, often 1-3 eV) | Appearance of new peaks/shoulders; clear chemical state change. |
| Dissociative Chemisorption | Bond breaking & new bond formation | Large shifts for both fragments | New species identified; stoichiometry changes. |
Table 2: Quantitative Detection Limits & Precision in Adsorption Studies
| Parameter | Typical Range in Adsorption Studies | Instrumental/Experimental Factor |
|---|---|---|
| Detection Sensitivity | ~0.1 - 1.0 at.% (monolayer sensitivity) | Analyzer transmission, X-ray flux, cross-section. |
| Binding Energy Precision | ±0.05 - 0.1 eV | Charge referencing, spectrometer calibration. |
| Depth Resolution | 1.5 - 5 nm (varies with take-off angle) | Photoelectron kinetic energy, analysis angle. |
| Spatial Resolution (Micro-XPS) | 5 - 30 µm | Focused X-ray probe size. |
Detailed Experimental Protocols
Protocol 1: Differentiating Physisorbed vs. Chemisorbed Protein Layers
- Objective: To determine the adsorption mechanism of a protein (e.g., Albumin) on a TiO₂ surface.
- Materials: TiO₂ substrate, Protein solution (1 mg/mL in PBS), XPS system with monochromatic Al Kα source.
- Procedure:
- Sample Preparation: Immerse TiO₂ substrate in protein solution for 2 hours at 25°C. Rinse gently with buffer (3x) to remove loosely bound (physisorbed) molecules. Dry under N₂ stream.
- XPS Data Acquisition:
- Mount sample using conductive tape.
- Acquire survey spectrum (0-1100 eV, Pass Energy 150 eV).
- Acquire high-resolution spectra for Ti 2p, O 1s, N 1s, and C 1s (Pass Energy 20-50 eV, ≥5 scans).
- Charge Reference: Adventitious C 1s set to 284.8 eV.
- Data Analysis:
- Quantify atomic % of N 1s (protein marker).
- Deconvolve Ti 2p spectrum. A decrease in Ti signal intensity indicates coverage.
- Analyze O 1s spectrum: A new component at ~531.5 eV may indicate Ti–O–C bond formation (chemisorption).
- Analyze N 1s spectrum: A single peak at ~399.8 eV suggests amine/amide (physisorption/H-bonding). A shift to ~398.5 eV may indicate direct N–Ti coordination (chemisorption).
Protocol 2: In-situ or Post-reaction Analysis of Catalytic Gas Adsorption
- Objective: To identify chemisorbed intermediates from CO on a Pt/γ-Al₂O₃ catalyst.
- Materials: Catalyst pellet, In-situ cell or glove bag for air-free transfer.
- Procedure:
- Pre-treatment & Adsorption: Reduce catalyst in H₂ at 300°C for 1 hour. Cool, expose to 1 bar CO at 25°C for 30 minutes. Purge with He.
- Sample Transfer: Use an air-free transfer vessel to move sample into XPS without air exposure.
- XPS Data Acquisition:
- Use a charge neutralizer (flood gun) for insulating catalyst support.
- Acquire high-resolution C 1s spectrum with high signal-to-noise.
- Data Analysis: Deconvolve C 1s spectrum. Peak assignments:
- ~284.8 eV: Adventitious carbon / C–C.
- ~285.8-286.2 eV: C–O (physisorbed/weakly bound).
- ~287.0-287.5 eV: Carbonyl (C=O) in adsorbed CO (chemisorbed).
- ~289.0+ eV: Carbonate species (dissociative chemisorption).
Visualization of Workflows and Relationships
Title: XPS Differentiation of Physisorption vs Chemisorption
Title: Standard XPS Protocol for Adsorption Analysis
The Scientist's Toolkit: Key Research Reagent Solutions & Materials
Table 3: Essential Materials for XPS Adsorption Studies
| Item | Function & Relevance to Adsorption Analysis |
|---|---|
| Monocrystalline Substrate (e.g., Au(111), Si Wafer) | Provides an atomically clean, flat, and well-defined surface for model adsorption studies. Essential for quantifying coverage and binding geometry. |
| High-Purity Gases (e.g., CO, O₂, H₂, Ultra-high Purity N₂) | Used for in-situ adsorption, pre-treatment (reduction, oxidation), and sample transfer. Critical for studying catalytic or sensor-related adsorption. |
| Charge Neutralization Flood Gun (Electron/ Ion) | Compensates for surface charging on insulating samples (e.g., polymers, oxides), ensuring accurate binding energy measurement for adsorbates. |
| In-situ Cell or Environmental Transfer Holder | Allows sample pre-treatment (heating, gas exposure) and transfer to the XPS analyzer under vacuum/controlled atmosphere, preserving adsorbed species. |
| Certified XPS Reference Materials (e.g., Au, Cu, Ag foils) | Used for regular energy scale calibration of the spectrometer, ensuring precision in detecting small BE shifts indicative of chemisorption. |
| Sputter Ion Gun (Ar⁺, C₆₀⁺) | Cleans the substrate surface prior to adsorption experiments and performs depth profiling to assess adsorbate layer thickness and uniformity. |
| Ultra-Pure Solvents (e.g., Ethanol, Acetone, Toluene) | Used for substrate cleaning and for preparing solutions of molecular adsorbates (e.g., polymers, biomolecules, organic catalysts). |
Within the broader thesis on X-ray Photoelectron Spectroscopy (XPS) analysis for distinguishing physisorbed and chemisorbed species, this document provides essential theoretical and practical guidelines. Accurate interpretation of binding energy (BE) shifts is fundamental for determining adsorption mechanisms, surface reactivity, and molecular orientation in materials science and drug development (e.g., drug-surface interactions, nanoparticle functionalization). This note consolidates expected BE shifts for common bonding types and provides protocols for their experimental verification.
Theoretical Framework: Expected Binding Energy Shifts
The core principle is that the measured BE of an electron is influenced by the chemical environment of the atom. A shift to higher BE (positive shift) indicates an increase in oxidation state or bonding to a more electronegative species. A shift to lower BE (negative shift) suggests a reduction in oxidation state or bonding to a more electropositive species.
Table 1: Expected XPS Binding Energy Shifts for Common Elements and Bonding Types
| Element (Core Level) | Bonding Type / Chemical State | Typical BE Range (eV) | Approx. Shift from Reference (eV) | Primary Cause of Shift |
|---|---|---|---|---|
| Carbon C 1s | C-C/C-H (aliphatic, adventitious) | 284.8 - 285.0 | 0.0 (Reference) | Reference standard. |
| C-O (alcohol, ether) | 286.1 - 286.9 | +1.3 to +2.1 | Bonding to electronegative O. | |
| C=O (carbonyl) | 287.2 - 288.2 | +2.4 to +3.4 | Increased oxidation state. | |
| O=C-O (carboxylate, ester) | 288.5 - 289.2 | +3.7 to +4.4 | Two electronegative O atoms. | |
| CF₂ (in polymers) | 291.0 - 291.5 | +6.2 to +6.7 | Extreme electronegativity of F. | |
| Nitrogen N 1s | Amine / -NH₂ | 398.7 - 399.5 | -- | Depends on protonation state. |
| Amide / -N-(C=O) | 399.5 - 400.3 | ~+0.5 to +1.0 vs amine | Electron withdrawal by carbonyl. | |
| Protonated Amine (-NH₃⁺) | 401.0 - 402.5 | +1.5 to +3.0 vs amine | Positive formal charge. | |
| Pyridinic N | 398.0 - 399.0 | -- | Aromatic, sp² hybridized. | |
| Oxygen O 1s | Metal Oxide (O²⁻) | 529.0 - 530.5 | -- | Lattice oxygen in metal oxides. |
| C=O (carbonyl) | 530.8 - 531.5 | -- | Distinguishable from O-C. | |
| C-O (hydroxyl, ether) | 532.2 - 533.0 | +1.0 to +2.0 vs metal oxide | Different bond polarity. | |
| Adsorbed H₂O / -OH | 532.5 - 533.5 | -- | High BE due to hydrogen bonding. | |
| Sulfur S 2p | Sulfide (S²⁻, e.g., in CdS) | 160.5 - 161.5 | -- | Reduced sulfur. |
| Organic Thiol / R-SH | 163.0 - 164.0 (S 2p₃/₂) | ~+2.0 vs sulfide | Covalent bonding to carbon. | |
| Sulfoxide (R-SO-R) | ~166 eV | +5.0 vs sulfide | Higher oxidation state (S=O). | |
| Sulfone (R-SO₂-R) | ~168 eV | +7.5 vs sulfide | Highest oxidation state (O=S=O). |
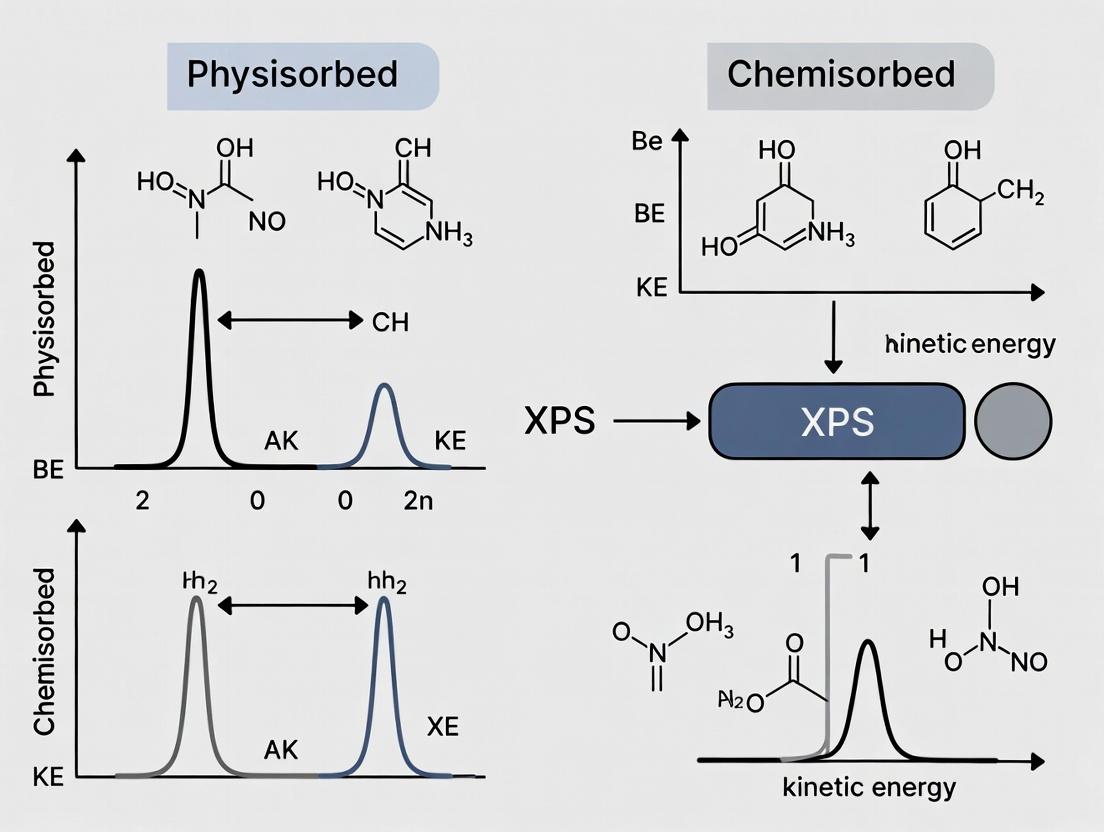
Table 2: Distinguishing Physisorption vs. Chemisorption via XPS Shifts
| Feature | Physisorption (Weak, van der Waals) | Chemisorption (Strong, Covalent/Ionic) |
|---|---|---|
| BE Shift Magnitude | Typically very small (< 0.5 eV). Often just a broadening of substrate signal. | Significant, measurable shifts (> 0.5 eV, often 1-4 eV). New distinct peaks may form. |
| Peak Shape | Asymmetric tailing or weak shoulder on main peak. | New, symmetric component that can be fitted separately. |
| Dose/Time Dependence | BE and intensity change linearly with exposure, may saturate due to monolayer formation. | Shows rapid initial shift then stabilization, indicative of bond formation. |
| Thermal Stability | Peaks diminish or disappear upon mild heating (e.g., to room temp or slightly above). | Peaks persist or transform (e.g., decompose) upon heating to higher temperatures. |
Experimental Protocols
Protocol 1: Sample Preparation for Adsorption Studies
Aim: To prepare a clean, well-defined substrate for the controlled adsorption of target species (e.g., drug molecules, functionalizing agents).
- Substrate Selection: Use a polished single crystal (e.g., Au(111), SiO₂ wafer) or a clean foil (e.g., Au, Ti) depending on the study.
- Cleaning: For metals, use cycles of Ar⁺ sputtering (1-2 keV, 5-15 µA, 5-10 min) followed by annealing (to restore crystallinity). For oxides, use solvent cleaning (sonication in acetone, isopropanol) followed by UV-Ozone treatment for 20-30 minutes to remove organics.
- In-Situ Adsorption: Transfer the clean substrate to an ultra-high vacuum (UHV) preparation chamber. Introduce the adsorbate via a leak valve (for gases) or using a precision doser (for vapors from liquids/solids). Exposure is given in Langmuirs (1 L = 10⁻⁶ Torr·sec).
- Ex-Situ Adsorption: For solutions (common in drug research), immerse the substrate in a controlled-concentration solution of the adsorbate for a set time. Rinse gently with a pure solvent to remove physisorbed multilayers and dry under a gentle N₂ stream.
Protocol 2: XPS Data Acquisition for Fingerprint Analysis
Aim: To acquire high-quality spectra enabling precise determination of BE shifts.
- Instrument Setup: Use a monochromatic Al Kα X-ray source (1486.6 eV). Set the pass energy to 20-50 eV for high-resolution regional scans.
- Charge Neutralization: For insulating samples (e.g., oxides, organic layers), use a low-energy electron flood gun in combination with an Ar⁺ screen to achieve stable, neutralized spectra.
- Energy Calibration: Reference all spectra to a known signal. For adventitious carbon, set the C 1s (C-C/H) peak to 284.8 eV. For clean metals, use the Fermi edge or a known metal peak (e.g., Au 4f₇/₂ at 84.0 eV).
- Data Collection: Acquire survey spectra (0-1100 eV) to identify all elements. Then acquire high-resolution spectra of all relevant core levels (e.g., C 1s, O 1s, N 1s, S 2p, substrate metal) with sufficient counts (>10k in the peak maximum).
- Angle-Resolved XPS (Optional): Vary the take-off angle (e.g., 90° = bulk-sensitive, 20° = surface-sensitive) to probe stratification of physisorbed vs. chemisorbed layers.
Protocol 3: Spectral Deconvolution and Shift Determination
Aim: To quantitatively analyze BE shifts from acquired spectra.
- Background Subtraction: Apply a Shirley or Tougaard background to the high-resolution spectrum.
- Peak Fitting: Use a least-squares fitting algorithm. Use a mix of Gaussian-Lorentzian line shapes (GL(30) is common). Constrain spin-orbit doublets (e.g., S 2p, Au 4f) with the correct splitting and area ratio.
- Component Assignment: Assign fitted components based on BE positions from Table 1. The number of components should be chemically justifiable.
- Shift Calculation: Report the BE of each component. The shift (ΔBE) is calculated relative to the appropriate reference (e.g., substrate peak, adventitious carbon at 284.8 eV, or a known internal standard).
- Error Reporting: State the estimated error in BE determination (typically ±0.1 eV for well-calibrated spectra on conductors, ±0.2 eV for insulators).
Visualization: Workflow & Data Interpretation
Diagram Title: XPS Workflow for Bonding Type Analysis
The Scientist's Toolkit: Research Reagent Solutions & Materials
Table 3: Essential Materials for XPS Adsorption Studies
| Item | Function & Explanation |
|---|---|
| Monocrystalline Substrates (Au(111), SiO₂/Thermal Oxide) | Provide atomically flat, chemically defined surfaces essential for fundamental adsorption studies and as a calibration reference. |
| High-Purity Solvents (HPLC Grade Acetone, Isopropanol, Water) | Used for ex-situ sample cleaning and preparation to prevent contamination that would obscure XPS signals from target adsorbates. |
| Ultra-High Purity Gases (Argon 99.999%, Nitrogen 99.999%) | Argon is for ion sputter cleaning. Nitrogen is for drying and creating inert atmospheres during sample transfer. |
| Precision Leak Valve & Gas Dosing System | Allows controlled introduction of gaseous adsorbates (e.g., O₂, CO, drug vapors) into UHV chambers for in-situ exposure studies. |
| Standard Reference Materials (Sputtered Au Foil, Clean Si Wafer) | Critical for daily verification of spectrometer energy scale and resolution performance. |
| Charge Neutralization Sources (Low-Energy e⁻ Flood Gun, Ar⁺ Screen) | Mandatory for analyzing insulating samples (most physisorbed/chemisorbed layers on oxides) to obtain reliable, non-shifted BEs. |
| Certified XPS Sensitivity Factor Database | Provided by spectrometer manufacturer; enables quantitative atomic concentration calculations from peak areas. |
| Spectral Database Software (e.g., NIST XPS Database, Commercial Libraries) | Contains collections of BE values for pure compounds, aiding in the initial assignment of unknown spectral features. |
Within the broader thesis on X-ray Photoelectron Spectroscopy (XPS) analysis for distinguishing physisorbed and chemisorbed species, understanding common physisorbed contaminants is critical. In biomedical surface analysis, physisorbed layers of adventitious carbon, water, and processing contaminants ubiquitously mask the true chemical state of materials, impacting data interpretation for biomaterials, implant surfaces, and drug delivery systems. This document provides application notes and detailed protocols for their identification and management.
Quantitative Data on Common Physisorbed Species
Table 1: Key Characteristics and XPS Signatures of Common Physisorbed Contaminants
| Contaminant Species | Typical XPS Binding Energy (C 1s / O 1s) | Common Source | Approximate Layer Thickness (on inert substrates) | Impact on Biomedical Surface Analysis |
|---|---|---|---|---|
| Adventitious Carbon (Hydrocarbons) | C-C/C-H: 284.8 - 285.0 eV; C-O: ~286.5 eV | Ambient air exposure, handling | 1 - 3 nm | Masks substrate signals; reference peak for charge correction. |
| Physisorbed Water (H₂O) | O 1s: ~533.0 eV (H₂O) vs. ~531.5 eV (O²⁻/OH⁻) | Humidity, sample prep | Monolayer to multilayers | Alters oxygen speciation; can obscure metal oxide/hydroxide signals. |
| Silicones (PDMS) | Si 2p: ~102 eV (SiOₓCᵧ); C 1s: ~284.8 & 286.5 eV | Lubricants, seals, tubing | Variable, often patchy | Causes significant C and Si signals; common in processed devices. |
| Phthalates & Plasticizers | C 1s: 284.8 eV (C-C), 286.5 eV (C-O), 289.0 eV (O=C-O) | Plastic containers, tubing | Nanometer-scale films | Introduces ester signatures; can interfere with polymer degradation studies. |
| Perfluorinated Compounds (PFCs) | C 1s: ~292 eV (CF₂), ~294 eV (CF₃); F 1s: ~689 eV | Anti-stick coatings, some labware | Sub-monolayer | Provides intense, high-BE carbon signals; contaminates sputter sources. |
Table 2: Recommended XPS Acquisition Parameters for Differentiating Physisorption vs. Chemisorption
| Parameter | Setting for Survey Scans | Setting for High-Resolution Scans | Rationale |
|---|---|---|---|
| Pass Energy | 100 - 150 eV | 20 - 50 eV | Balances sensitivity and resolution for detection/quantification. |
| Step Size | 1.0 eV | 0.05 - 0.1 eV | Necessary to resolve subtle shifts from physisorbed vs. chemisorbed O/C states. |
| Charge Neutralization | Always ON | Always ON | Essential for insulating biomedical samples (polymers, oxides). |
| Analysis Area | 500 µm x 500 µm (min) | 200 µm x 200 µm | Larger area averages heterogeneous contamination. |
| Take-off Angle (for depth sensitivity) | 90° (normal) | 90° or 30° (grazing) | Grazing angle increases surface sensitivity for monolayer contaminants. |
Experimental Protocols
Protocol 1: Minimizing and Characterizing Adventitious Carbon for Reference Surface Generation
Objective: Prepare a clean gold substrate to assess adventitious carbon deposition rate under controlled conditions. Materials: Ultra-flat gold-coated wafer, UHV transfer vessel, solvents (see Toolkit). Procedure:
- Solvent Cleaning: Sonicate gold substrate in ACS-grade toluene for 10 minutes, followed by acetone for 10 minutes, and finally isopropanol for 10 minutes.
- Drying: Dry under a stream of Argon or Nitrogen gas (99.999% purity).
- Immediate Transfer: Load sample into a nitrogen-purged transfer vessel within 60 seconds of drying.
- UHV Introduction: Introduce the sample into the XPS load lock within 15 minutes. Pump down to ≤1×10⁻⁸ mbar.
- XPS Analysis:
- Acquire a survey scan (0-1200 eV) immediately upon entry into analysis chamber.
- Acquire high-resolution scans of C 1s, O 1s, and Au 4f regions.
- Use the Au 4f₇/₂ peak at 84.0 eV as an internal reference. The C 1s hydrocarbon peak (C-C/C-H) is set to 284.8 eV for charge referencing on other materials.
- Controlled Exposure: Vent the load lock with dry, hydrocarbon-filtered nitrogen for a set time (e.g., 5 min). Re-pump and re-analyze to measure carbon buildup.
Protocol 2: Differentiating Physisorbed Water from Hydroxyl Groups on Titanium Implant Alloys
Objective: Use in-situ temperature control and angle-resolved XPS to distinguish H₂O from Ti-OH. Materials: Cleaned Ti-6Al-4V coupon, UHV-compatible heating/cooling stage. Procedure:
- Initial Characterization: Insert cleaned sample. Acquire high-resolution O 1s spectrum at -120°C (cooled via stage) to freeze physisorbed water.
- Spectral Deconvolution: Fit the O 1s peak with three components:
- O²⁻ in TiO₂ (~530.0 eV)
- OH⁻ (chemisorbed hydroxyl) (~531.5 eV)
- H₂O (physisorbed) (~533.0 eV)
- Temperature-Dependent Desorption: Gradually warm the sample to 25°C, then to 150°C, acquiring O 1s spectra at 25°C intervals.
- Data Analysis: Plot the area of the ~533.0 eV component versus temperature. A sharp decrease between -50°C and 50°C confirms physisorbed water. The persistent ~531.5 eV component indicates chemisorbed hydroxyls.
Protocol 3: Identifying Silicone Contamination on Drug-Eluting Polymer Films
Objective: Detect trace physisorbed polydimethylsiloxane (PDMS) from processing equipment. Materials: Poly(lactic-co-glycolic acid) (PLGA) film, argon gas cluster ion source (optional). Procedure:
- Standard XPS: Acquire survey and high-resolution scans of C 1s, O 1s, and Si 2p regions.
- Spectral Analysis: Look for doublet in Si 2p region at ~102 eV (Si 2p₃/₂) and ~102.7 eV (Si 2p₁/₂), indicative of SiOₓCᵧ.
- Gentle Surface Cleaning (if required): Use a low-energy (≤2 kV) Argon Gas Cluster Ion Beam (GCIB) for 30 seconds to remove physisorbed layer without damaging underlying PLGA.
- Post-Cleaning Analysis: Re-acquire Si 2p spectrum. Significant reduction of Si signal confirms physisorption; persistent signal suggests harder-to-remove contamination or incorporated silicone.
Visualization: Pathways and Workflows
Title: Sources and Impact of Physisorbed Contaminants on XPS Analysis
Title: Experimental Workflow for Physisorbed Species Identification
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Materials for Managing Physisorbed Contaminants in XPS Analysis
| Item | Function & Relevance | Example Product/Criteria |
|---|---|---|
| Solvent Series (Toluene, Acetone, IPA) | Sequential removal of organic processing residues and grease. | ACS Grade or better, in glass bottles with low extractables. |
| Dry, Hydrocarbon-Filtered Nitrogen Gas | For sample drying and creating minimal-contamination environments. | 99.999% purity, with inline hydrocarbon/moisture trap. |
| Ultra-High Vacuum (UHV) Transfer Vessel | Allows sample movement from glovebox to XPS without air exposure. | Metal-sealed, capable of maintaining <1×10⁻⁸ mbar. |
| UHV-Compatible Heating/Cooling Stage | For in-situ temperature-dependent studies to desorb water/volatiles. | Range: -120°C to +500°C, with precise temperature control. |
| Argon Gas Cluster Ion Source (GCIB) | For gentle removal of physisorbed layers without damaging delicate polymers. | Cluster size: 1000-5000 atoms, energy <10 keV. |
| Standard Reference Substrates | For adventitious carbon reference and instrument function checks. | Sputter-cleaned gold foil, highly ordered pyrolytic graphite (HOPG). |
| Charge Neutralization Flood Gun | Essential for analyzing insulating biomedical samples (polymers, ceramics). | Low-energy electron flood gun combined with low-energy ion flood. |
Application Notes
The study of chemisorbed species—where adsorbates form strong, directed chemical bonds with a substrate—is central to advancing surface science and applied materials research. In the broader thesis investigating X-ray Photoelectron Spectroscopy (XPS) for differentiating physisorbed and chemisorbed states, these three classes represent critical, high-impact case studies. XPS provides quantitative data on elemental composition, chemical state, and binding energy shifts, allowing researchers to confirm covalent attachment, assess layer integrity, and monitor reaction efficacy. This document outlines protocols and application notes for the XPS-assisted analysis of functionalized self-assembled monolayers (SAMs), covalently tethered drug molecules, and bifunctional covalent linkers.
Functionalized Layers (e.g., Silane & Thiol SAMs)
Functionalized layers serve as the foundational chemisorbed interface for subsequent immobilization. Organosilanes on oxides and alkanethiols on gold are quintessential systems.
XPS Insights: Successful chemisorption is indicated by the appearance of substrate-specific signals (e.g., Si 2p for silanes on silicon wafer, S 2p for thiols on Au) and the expected elemental ratios from the functional headgroup (e.g., N 1s for an amine-terminated SAM). A key metric is the attenuation of the underlying substrate signal, which correlates with monolayer thickness and density.
Table 1: XPS Binding Energy Signatures for Common Functionalized Layers
| Layer System | Core Level | Binding Energy (eV) | Chemical State Indication |
|---|---|---|---|
| APTES on SiO₂ | Si 2p | 102.2 | C-Si-O (Siloxane) |
| N 1s | 399.5 | -NH₂ | |
| O 1s | 532.8 | Si-O-Si, Si-O-C | |
| Octadecanethiol on Au | S 2p | 162.0 (2p₃/₂) | Au-S (Thiolate) |
| C 1s | 284.8 | C-C/C-H | |
| Au 4f | 84.0 (4f₇/₂) | Attenuated Substrate | |
| PEG-Thiol on Au | O 1s | 532.5 | C-O-C |
| C 1s | 286.5 | C-O |
Covalently Tethered Drug Molecules
Covalent immobilization of drug molecules (e.g., kinase inhibitors, antibiotics) onto biomedical device surfaces or drug delivery nanoparticles requires robust chemisorption to ensure localized, sustained action.
XPS Insights: The analysis verifies the drug's unique elemental "fingerprint" (e.g., F 1s in fluorinated drugs, specific N 1s environments in heterocycles) on the surface. Quantification of these species relative to the linker atoms confirms grafting density. Control experiments with physisorbed drugs show significantly attenuated signals after rigorous solvent washing, while chemisorbed layers remain stable.
Table 2: XPS Analysis of Covalently Immobilized Drug Molecules
| Drug / Target | Key XPS Elemental Marker | Typical BE Range (eV) | Protocol for Confirmation |
|---|---|---|---|
| Doxorubicin (Anthracycline) | N 1s | 399.8 | N/C ratio increase post-conjugation vs. linker alone |
| Ciprofloxacin (Quinolone) | F 1s | 688.5 | Appearance of F signal on non-fluorinated substrate |
| Gefitinib (Kinase Inhibitor) | N 1s, F 1s | N: 399.5; F: 687.8 | High-resolution scan of N 1s to deconvolute multiple species |
Covalent Linkers (Heterobifunctional Crosslinkers)
Linkers like NHS esters, maleimides, and click chemistry reagents (e.g., DBCO, azides) form the critical chemisorbed bridge between surfaces and biomolecules.
XPS Insights: XPS tracks the consumption of one reactive group and the introduction of new elements from the coupled molecule. For example, the decrease in a NHS ester's O 1s component (ester oxygen) and the concurrent rise in N 1s signal from an amide bond and the newly attached protein confirm successful linkage.
Table 3: XPS Tracking of Crosslinker Reaction Steps
| Linker/Reaction | Elemental Probe | Change Indicating Success |
|---|---|---|
| Sulfo-SMCC (NHS ester + Maleimide) | S 2p (sulfonate), N 1s | S signal stable (linker); N 1s amide peak appears post-protein coupling |
| DBCO-Azide Click | N 1s (azide) | Azide N 1s peak (~405 eV) diminishes, new triazole N peak (~401 eV) forms |
| APTES + Glutaraldehyde | N 1s, C 1s | New C 1s C=O component (~288 eV) after aldehyde activation |
Experimental Protocols
Protocol 1: XPS Analysis of Aminosilane (APTES) Monolayer Formation on Silicon Wafer
Objective: To form and characterize a chemisorbed amine-functionalized layer for subsequent bioconjugation.
Materials:
- Piranha-cleaned silicon wafer (SiO₂ native oxide)
- (3-Aminopropyl)triethoxysilane (APTES)
- Anhydrous toluene
- Ethanol, HPLC grade
- Nitrogen stream
Procedure:
- Substrate Cleaning: Treat Si wafer in piranha solution (3:1 H₂SO₄:H₂O₂) for 30 min. CAUTION: Highly exothermic and corrosive. Rinse copiously with Milli-Q water and dry under N₂ stream. Perform immediate XPS survey scan to confirm clean surface (Si, O, C only; C contamination minimal).
- Silane Solution Preparation: In a glove box or under dry N₂, prepare a 2% (v/v) solution of APTES in anhydrous toluene.
- SAM Formation: Immerse the clean wafer in the APTES solution for 2 hours at room temperature in a sealed, dry environment.
- Washing: Remove wafer and sonicate sequentially in toluene (2 min), ethanol (2 min), and fresh ethanol (2 min) to remove physisorbed silane.
- Curing: Bake wafer at 110°C for 10 min to promote siloxane (Si-O-Si) network formation.
- XPS Characterization:
- Survey Scan: Confirm presence of Si, O, C, and N.
- High-Resolution Scans: Acquire Si 2p, O 1s, C 1s, and N 1s regions.
- Data Analysis: Quantify atomic percentages. The Si 2p peak should show a component at ~102.2 eV (Si-C from APTES). The N 1s peak at ~399.5 eV confirms primary amine presence. Compare C/Si and N/Si ratios to theoretical monolayer values.
Protocol 2: Immobilization of a Drug Molecule via EDC/NHS Coupling and XPS Verification
Objective: To covalently attach doxorubicin (DOX) to a carboxyl-terminated SAM and verify chemisorption via XPS.
Materials:
- COOH-terminated alkanethiol SAM on Au (from Protocol 1 analogue)
- Doxorubicin hydrochloride
- EDC (1-Ethyl-3-(3-dimethylaminopropyl)carbodiimide)
- Sulfo-NHS (N-Hydroxysulfosuccinimide)
- MES buffer (0.1 M, pH 5.5) and PBS (pH 7.4)
Procedure:
- Surface Activation: Incubate the COOH-SAM substrate in a freshly prepared solution of 50 mM EDC and 25 mM Sulfo-NHS in MES buffer for 30 min at RT. Rinse gently with MES buffer.
- Drug Coupling: Immediately transfer the substrate to a 1 mM solution of DOX in PBS (pH 7.4). Incubate for 2 hours in the dark at RT.
- Control Sample: Prepare a physisorption control by incubating a COOH-SAM in 1 mM DOX without EDC/NHS activation.
- Washing: Wash both samples vigorously by agitation in PBS, Milli-Q water, and ethanol (3x each for 5 min) to remove non-covalently bound drug.
- XPS Characterization:
- Survey Scan: Note key DOX markers: N (from the sugar amine) and unique trace elements if present (e.g., Cl from HCl salt).
- High-Resolution C 1s & N 1s: Deconvolute C 1s to identify amide bond component (~288.0 eV). The N 1s should show a clear peak (~399.8 eV) on the covalently grafted surface.
- Quantification: The N/Au atomic ratio will be significantly higher for the chemisorbed sample compared to the washed physisorption control, confirming successful covalent linkage.
Diagrams
Title: XPS Workflow for Chemisorbed Species Analysis
Title: Covalent Drug Immobilization Pathway
The Scientist's Toolkit
Table 4: Essential Research Reagent Solutions for Chemisorption Studies
| Reagent / Material | Primary Function | Key Consideration for XPS Analysis |
|---|---|---|
| Piranha Solution (H₂SO₄:H₂O₂) | Ultimate oxidizer for cleaning glass, Si, Au surfaces. Removes organic residue. | Provides ultra-clean, high-surface-energy substrate for reproducible SAM formation. Essential for low initial C 1s signal. |
| Anhydrous Toluene | Solvent for preparing silane solutions. | Anhydrous conditions prevent silane polymerization in solution, ensuring monolayer rather than multilayer deposition. |
| Alkanethiols (e.g., 16-Mercaptohexadecanoic acid) | Forms chemisorbed SAM on Au, Ag, Pt. Terminal group (-COOH, -OH, -NH₂) provides functionality. | Chain length affects SAM order and XPS photoelectron attenuation. S 2p peak confirms thiolate bond. |
| Organosilanes (e.g., APTES, GPTMS) | Forms chemisorbed layer on hydroxylated oxides (SiO₂, TiO₂). | Requires surface hydroxyl groups. XPS monitors Si 2p shift from substrate SiO₂ to siloxane. |
| EDC / Sulfo-NHS | Carbodiimide crosslinker system for activating carboxyl groups to form amide bonds with amines. | Reaction efficiency is monitored by the appearance of the amide N 1s and C 1s peaks. |
| Heterobifunctional Linkers (e.g., Sulfo-SMCC) | Contains two different reactive groups (e.g., NHS ester & maleimide) for orthogonal, stepwise conjugation. | XPS can track sulfur species (sulfonate vs. maleimide) and nitrogen species to confirm each reaction step. |
Practical XPS Protocols: From Sample Prep to Peak Deconvolution for Adsorbed Layers
Best Practices for Sample Preparation and Handling to Preserve Surface State
Within the broader research on physisorbed and chemisorbed species using X-ray Photoelectron Spectroscopy (XPS), the integrity of the sample surface state is paramount. The measured signals, particularly for weakly bound physisorbed layers or specific oxidation states of chemisorbed species, are exquisitely sensitive to contamination and unintended modification introduced during handling. This document outlines critical protocols to preserve the relevant surface chemistry from the point of sample generation to insertion into the XPS instrument.
The following table summarizes major contamination sources and their documented effects on surface-sensitive measurements.
Table 1: Primary Contamination Sources and Their Impact on Surface Analysis
| Contamination Source | Example | Typical Quantified Impact on XPS Signals | Effect on Physisorbed/Chemisorbed Species |
|---|---|---|---|
| Atmospheric Exposure | O₂, H₂O, Organic Vapors | Carbonaceous layer growth: 0.2 - 0.5 nm/min initial rate. Adventitious Carbon (C-C/C-H) signal can dominate spectrum in <5 min. | Physisorbed H₂O/O₂ can replace or mask target species. Can induce oxidation, altering chemisorbed state. |
| Direct Contact | Gloves, Tools, Packaging | Salt (Na, K) transfer from gloves: Atomic concentration can increase by 1-5%. Silicone transfer is common. | Complete displacement or mixing of physisorbed layer. Contamination peaks obscure elemental/chemical state regions. |
| Outgassing | Adhesives, Polymers, Samples | Hydrocarbon background rise in chamber pressure (>1x10⁻⁸ mBar). Contaminates analysis chamber. | Can deposit a new, non-representative physisorbed layer in vacuo. |
| Improper Cleaning | Solvent Residue, Lint | Solvent (e.g., acetone) residue can add 3-8% oxygen signal. Lint adds cellulose (C-O) signal. | Dissolves or reacts with target species. Adds interfering chemical states. |
| Electrostatic & Thermal Damage | Charging, Local Heating | Can shift apparent binding energy by several eV. May cause reduction/desorption. | Desorption of physisorbed species. Beam-induced reactions in chemisorbed layer. |
Experimental Protocols
Protocol 1: Inert Atmosphere Transfer for Air-Sensitive Samples
Objective: To transfer a freshly prepared sample from a glovebox (or reaction chamber) to the XPS load lock without atmospheric exposure.
- Materials: Argon-filled glovebox (<1 ppm O₂/H₂O), antechamber/transfer vessel, XPS with load lock compatible with transfer vessel.
- Procedure: a. Prepare sample inside the inert atmosphere glovebox. b. Secure the sample on a dedicated transfer holder. Do not use adhesive tapes. c. Place the holder inside a sealed transfer vessel within the glovebox. d. Evacuate the transfer vessel's antechamber (if applicable) or keep it permanently under inert gas purge. e. Dock the transfer vessel directly to the XPS load-lock port. f. Use the instrument's pumping sequence to introduce the sample into the ultra-high vacuum (UHV) environment.
Protocol 2: Minimal-Exposure Ex-Situ Preparation
Objective: To prepare samples that must be created outside UHV (e.g., solution-deposited films) with minimal contamination.
- Materials: Cleanroom wipes, analytical-grade solvents (e.g., HPLC-grade), stainless steel tweezers with smooth edges, nitrogen gun with particulate filter, clean glass Petri dishes.
- Procedure: a. Substrate Cleaning: Clean substrate (e.g., Si wafer) via sonication in successive solvents (e.g., acetone, followed by isopropanol). Rinse with pure solvent and dry under a stream of filtered N₂. b. Sample Deposition: Perform deposition (e.g., spin-coating, drop-casting) in a laminar flow hood, if possible. c. Post-Processing: If drying is required, use a gentle, filtered N₂ stream in a covered but not sealed environment to prevent solvent condensation. d. Mounting: Using clean tweezers, mount the sample on the XPS holder. Avoid touching the analysis area. e. Immediate Transfer: Place the mounted sample into a sealed, inert container (e.g., jar purged with Ar) immediately. Label the container. f. Rapid Insertion: Transfer the container to the XPS lab and insert the sample into the load lock within 15 minutes of preparation.
Protocol 3: In-Situ Cleavage or Fracture
Objective: To generate a pristine surface within the UHV environment of the XPS system.
- Materials: XPS system with in-situ sample preparation chamber (UHV), fracture stage or cleaver, sample rods with appropriate fixtures.
- Procedure: a. Mount the bulk crystal or material onto the in-situ cleaving stage following manufacturer instructions. b. Transfer the sample into the UHV preparation chamber. Pump down to base pressure (<5x10⁻⁹ mBar). c. Using a wobble stick or internal mechanism, actuate the cleaver to fracture the sample along its crystal plane or a desired interface. d. Immediately transfer the freshly cleaved sample to the analysis position without breaking vacuum. e. Begin spectral acquisition as soon as possible to monitor surface stability.
The Scientist's Toolkit
Table 2: Essential Research Reagent Solutions for Surface-Preserving Preparation
| Item | Function & Rationale |
|---|---|
| Stainless Steel Tweezers (Flat-Blade) | For sample handling; less likely to shed particles or transfer organics compared to coated tweezers. Can be cleaned by solvent and plasma. |
| HPLC/ACS Grade Solvents | High-purity solvents minimize non-volatile residue left on the sample after cleaning or processing. |
| Particulate-Filtered Nitrogen Gun | Provides a clean, dry gas stream for drying samples without introducing oil droplets or particles. |
| Argon-Filled Glovebox | Provides inert atmosphere for synthesis, preparation, and mounting of highly air/water-sensitive materials. |
| Indium Foil (High Purity) | A soft, conductive metal used to mount powdered samples without chemical adhesive, improving electrical contact and minimizing contamination. |
| UHV-Compatible Conductive Tape (e.g., Carbon) | Used for mounting when necessary; selected for low outgassing properties to maintain chamber pressure. |
| In-Situ Cleaving/Fracture Stage | Integrated into the XPS vacuum system to expose bulk interfaces or pristine surfaces without air exposure. |
| Antistatic Gun | Neutralizes static charge on insulating samples prior to insertion, preventing particulate attraction and minimizing charging during analysis. |
Workflow Diagrams
Title: Sample Preparation Pathway Decision Tree
Title: Contamination Source to Mitigation Logic Chain
Within the broader thesis investigating X-ray Photoelectron Spectroscopy (XPS) for distinguishing and analyzing physisorbed and chemisorbed species, the selection of instrumental parameters is critical. Sensitive layers, such as self-assembled monolayers, organic thin films, or adsorbed biomolecules, are susceptible to beam damage and often yield weak signals. This application note provides a detailed protocol for selecting Pass Energy, Dwell Time, and Spot Size to maximize signal-to-noise ratio (SNR) and minimize radiation damage, ensuring accurate data for surface chemical state analysis in fields like drug delivery system characterization and biosensor development.
Core Parameter Optimization for Sensitive Layers
Pass Energy
Pass Energy (PE) controls the energy resolution and sensitivity of the analyzer. A lower PE yields higher energy resolution but lower sensitivity (count rate), and vice-versa. For sensitive, beam-sensitive layers, a balance must be struck.
- High-Resolution Spectra (for chemical state analysis): Use a lower PE (e.g., 10-50 eV). This is essential for resolving subtle shifts from chemisorbed species.
- Survey Spectra or Rapid Analysis: Use a higher PE (e.g., 80-160 eV) to increase throughput and reduce dose during initial assessment.
Recommendation: Acquire survey spectra at high PE, then acquire high-resolution regional scans at the lowest PE that provides an acceptable SNR within a tolerable acquisition time.
Dwell Time and Total Dose
Dwell time (time per data point) and total scan time directly influence radiation dose and signal quality. Excessive dose leads to degradation of physisorbed and many organic layers.
- Strategy: Use the minimum dwell time that provides a usable signal. It is often better to perform multiple short scans and co-add the data than one long, continuous scan, as this can allow for checking degradation.
- Protocol for Dose Management:
- Set a conservative initial dwell time (e.g., 50-100 ms).
- Acquire a rapid test scan.
- Increase dwell time iteratively only if the SNR is inadequate, monitoring for peak shape changes indicating damage.
Spot Size
The X-ray beam spot size determines the analyzed area and the spatial localization of the dose.
- Large Spot (e.g., 400-900 µm): Spreads the X-ray flux over a larger area, reducing the flux density (dose rate) on the sample. This is generally preferred for homogeneous, sensitive layers to minimize damage per unit area.
- Small Spot (e.g., 10-200 µm): Concentrates flux, increasing dose rate and risk of damage. Use only when spatial resolution is paramount (e.g., analyzing a specific feature), or if the layer is unexpectedly robust.
Quantitative Parameter Comparison Table
Table 1: XPS Parameter Guidelines for Sensitive Adsorbed Layers
| Parameter | Typical Range | Recommended for Sensitive Layers | Primary Trade-off | Impact on Physisorbed/Chemisorbed Studies |
|---|---|---|---|---|
| Pass Energy | 5-200 eV | Regional Scans: 20-50 eVSurvey Scans: 80-140 eV | Resolution vs. Sensitivity | High resolution (low PE) is critical to distinguish chemical state shifts of chemisorbed species. |
| Dwell Time | 10-1000 ms | 50-200 ms (start low) | SNR vs. Radiation Dose | Short dwell times and multiple scans help detect beam-induced desorption of physisorbed species. |
| Spot Size | 10-900 µm | ≥ 400 µm (if sample homogeneity allows) | Spatial Resolution vs. Flux Density | Larger spot reduces dose rate, preserving the integrity of weakly-bound physisorbed overlayers. |
| Number of Scans | 1-100+ | 5-20 (co-added) | Total Dose vs. SNR | Multi-scan co-addition allows monitoring of spectral changes (e.g., C-C/C-H attenuation) indicating damage. |
Detailed Experimental Protocols
Protocol A: Establishing a Damage Threshold for a New Sensitive Layer
Objective: Determine the maximum tolerable X-ray dose before significant sample degradation. Materials: The sample with the adsorbed sensitive layer, XPS system with monochromatic Al Kα source. Procedure:
- Initial Setup: Select a large spot size (e.g., 500 µm). Set the analyzer to a medium Pass Energy (e.g., 100 eV) for rapid data collection.
- Define Test Region: Choose a single, representative region on the sample.
- Sequential Spectral Acquisition:
- Acquire a high-resolution spectrum of a key element (e.g., C 1s, N 1s) using a low Pass Energy (e.g., 20 eV) and moderate dwell time (100 ms). Record the total acquisition time.
- Immediately acquire the same spectrum again under identical conditions.
- Repeat for 5-10 cycles.
- Data Analysis: Plot the intensity of characteristic peaks (e.g., C-C/C-H, C-O, C=O for organics) or the total area of a core level versus cumulative acquisition time (proportional to dose).
- Threshold Determination: The damage threshold is identified as the dose at which a statistically significant (>5%) decrease in the characteristic peak intensity or a change in the peak shape/fitting component ratio is observed. All subsequent experiments should use total doses below this threshold.
Protocol B: Optimized Data Acquisition for High-Resolution Chemical State Analysis
Objective: Acquire high-quality, high-resolution spectra of a sensitive adsorbed layer with minimal damage. Materials: Sample, XPS system. Procedure:
- Survey Scan: Using a large spot (500 µm), high Pass Energy (150 eV), and fast scan (low dwell time), collect a survey spectrum to identify all elements present.
- Parameter Set for Regional Scans:
- Spot Size: Maintain large spot (500 µm).
- Pass Energy: Set to 20-30 eV for optimal resolution/sensitivity balance.
- Dwell Time: Use the value determined from Protocol A, or start with 100 ms.
- Scan Regions: Define narrow energy windows for each core level of interest (e.g., ±10-15 eV around the peak center).
- Multi-Scan Co-addition: Program the system to acquire 5-10 scans of each region in immediate succession. Ensure the total estimated dose is below the damage threshold.
- Damage Monitoring: Compare the first and last co-added scan (or analyze a diagnostic peak like C 1s from a contaminant/adventitious carbon if the layer contains no carbon) for signs of damage (peak broadening, intensity loss, chemical shift).
- Data Processing: Co-add all scans from the sequence. Apply standard charge correction (e.g., referencing adventitious C 1s to 284.8 eV) and analyze using peak fitting procedures appropriate for chemisorbed species.
Experimental Workflow & Parameter Decision Logic
XPS Parameter Optimization Workflow for Sensitive Layers
The Scientist's Toolkit: Essential Research Reagents & Materials
Table 2: Key Materials for XPS Analysis of Adsorbed Species
| Item | Function in Research | Example/Notes |
|---|---|---|
| Monocrystalline Substrate (e.g., Au(111), Si(100)) | Provides an atomically flat, well-defined surface for adsorption studies, minimizing spectral broadening from substrate roughness. | Essential for model studies of chemisorption. |
| Self-Assembled Monolayer (SAM) Precursors (e.g., alkanethiols, silanes) | Model systems for creating well-ordered, chemisorbed layers of known thickness and chemistry. | Used to calibrate sensitivity factors and damage thresholds. |
| Ultra-Pure Solvents (e.g., HPLC-grade ethanol, toluene) | For cleaning substrates and preparing adsorption solutions without leaving contaminant residues. | Residual carbon can interfere with C 1s signals from the layer. |
| Plasma Cleaner (O₂/Ar) | Provides a reproducible method for generating atomically clean, hydrophilic substrate surfaces prior to adsorption. | Removes organic contaminants and activates oxide surfaces. |
| Inert Atmosphere Transfer Kit (Glove bag/box) | Allows transfer of air-sensitive samples (e.g., some physisorbed organics, reactive surfaces) into the XPS introduction chamber without air exposure. | Prevents oxidation or contamination before analysis. |
| Charge Neutralization System (Flood gun) | Essential for analyzing insulating sensitive layers (e.g., polymers, oxides) to prevent peak shifting and broadening due to surface charging. | Low-energy electrons/ions stabilize the surface potential. |
| Sputter Ion Source (Ar⁺) | Used cautiously for depth profiling or cleaning reference areas. Not typically used on the sensitive layer itself as it causes damage. | Can be used to clean substrate before adsorption. |
Acquiring High-Resolution Spectra for Key Elements (C 1s, O 1s, N 1s) to Probe Bonding
Within a broader thesis investigating XPS analysis for physisorbed and chemisorbed species, high-resolution spectral acquisition is paramount. Physisorption induces small, often sub-eV, binding energy (BE) shifts primarily through polarization, while chemisorption forms new chemical bonds, resulting in shifts of 1-3 eV or more. Deconvoluting high-resolution spectra of the C 1s, O 1s, and N 1s core levels is therefore critical to fingerprint surface composition, identify functional groups, and distinguish between adsorbed contaminants, intentionally grafted molecules, and substrate atoms in different bonding states. This protocol details the steps for acquiring publication-quality data to probe these subtle bonding differences.
Key Parameters for High-Resolution Spectra Acquisition
The following parameters, derived from current literature and instrumentation manuals, are optimized for a modern, monochromatic Al Kα X-ray source (1486.6 eV) spectrometer.
Table 1: Recommended Acquisition Parameters for High-Resolution Spectra
| Parameter | C 1s | O 1s | N 1s | Rationale |
|---|---|---|---|---|
| Pass Energy | 20-50 eV | 20-50 eV | 20-50 eV | Balances energy resolution and signal intensity. Lower values (20 eV) yield best resolution. |
| Step Size | 0.05-0.10 eV | 0.05-0.10 eV | 0.05-0.10 eV | Adequate sampling for narrow peaks (FWHM ~0.6-1.2 eV). |
| Dwell Time | 50-200 ms | 50-200 ms | 50-200 ms | Sufficient counts without excessive time; adjust based on sensitivity. |
| Number of Scans | 10-30 | 10-30 | 10-30 | Improves signal-to-noise ratio (SNR) for weak signals or dilute species. |
| Spot Size | 200-500 µm | 200-500 µm | 200-500 µm | Standard high-resolution area for homogeneous samples. |
| Charge Neutralizer | Always ON (for insulating samples) | Always ON (for insulating samples) | Always ON (for insulating samples) | Essential for accurate BE positioning on polymers, oxides, organics. |
Table 2: Reference Binding Energies for Common Functionalities
| Element | Functional Group / State | Approx. BE (eV) | Typical FWHM (eV) | Note |
|---|---|---|---|---|
| C 1s | C-C/C-H (adventitious, aliphatic) | 284.8 (Ref.) | 0.8-1.2 | Charge reference standard. |
| C-O (alcohol, ether) | 286.3-286.5 | 0.9-1.3 | Distinct in chemisorbed alkoxy groups. | |
| C=O (carbonyl) | 287.8-288.2 | 0.9-1.3 | Present in physisorbed adsorbates. | |
| O-C=O (ester, acid) | 288.8-289.2 | 0.9-1.3 | ||
| O 1s | Metal-O (oxide, hydroxide) | 529-531 | 1.0-1.5 | Broad, often asymmetric. |
| C=O (carbonyl) | 531.0-531.8 | 1.0-1.4 | Overlaps with many other species. | |
| C-O (alcohol, ether) | 532.5-533.2 | 1.0-1.4 | Distinguishing chemisorbed vs. physisorbed O is challenging. | |
| Adsorbed H₂O | 533.0-533.5 | 1.2-1.8 | Indicates physisorbed species. | |
| N 1s | Pyridinic / -N= | 398.5-399.0 | 0.9-1.3 | Common in N-doped materials. |
| Aminic / -NH- | 399.5-400.2 | 0.9-1.3 | From chemisorbed aminosilanes, peptides. | |
| Quaternary N⁺ | 401.0-402.5 | 0.9-1.3 | Protonated or methylated amine. | |
| N-O (nitro) | 405-407 | 1.0-1.5 | Oxidized nitrogen. |
Detailed Experimental Protocol
Protocol 1: Sample Preparation for Adsorbed Species Analysis
- Substrate Cleaning: Sonicate substrate (e.g., Au, Si, SiO₂, TiO₂) in sequential solvents (toluene, acetone, ethanol) for 10 minutes each. Dry under a stream of UHP N₂ or Ar.
- Adsorption: For physisorption, immerse the substrate in a dilute solution (0.1-1 mM) of the target molecule for 1-24 hours at room temperature. For chemisorption (e.g., SAM formation), use solutions with reactive headgroups (thiols, silanes) and control temperature/concentration per established literature.
- Rinsing: Rinse thoroughly with a pure solvent that dissolves the physisorbed but not the chemisorbed species (typically the same as the deposition solvent) to remove multilayers and weakly physisorbed molecules. Rinse 3-5 times.
- Drying: Dry under a gentle stream of UHP N₂. Avoid heating unless required for specific chemisorption.
- Transfer: Mount sample on a holder using double-sided conductive carbon tape. Transfer to XPS load-lock as swiftly as possible to minimize airborne hydrocarbon contamination.
Protocol 2: XPS Instrument Setup and Spectral Acquisition
- Initial Pump-Down: Evacuate load lock to ≤ 5 x 10⁻⁷ mBar before transferring to analysis chamber (≤ 1 x 10⁻⁹ mBar preferred).
- Charge Neutralization: For non-conductive samples, activate the low-energy electron flood gun and/or low-energy Ar ion flood. Optimize settings (typically 0.1-2 eV electrons) using a known standard to minimize peak broadening and shifting.
- Survey Scan: Acquire a wide survey spectrum (e.g., 0-1100 eV, 100 eV pass energy) to identify all elements present.
- High-Resolution Setup:
- Select the analytical area (200-500 µm spot).
- Set lens mode to "High Magnification" or "High Resolution."
- For each region (C 1s, O 1s, N 1s), input the parameters from Table 1. A typical sequence: C 1s → O 1s → N 1s.
- Energy Alignment & Referencing:
- Acquire the C 1s region first. Identify the dominant C-C/C-H peak from adventitious carbon.
- Set this peak's maximum to 284.8 eV using the spectrometer's software charge correction function.
- Apply this same correction offset to all subsequently acquired high-resolution spectra (O 1s, N 1s).
- Data Collection: Initiate automated scans. Monitor the SNR in real-time; increase scans if the signal for the species of interest (e.g., N 1s in a dilute layer) is poor.
Protocol 3: Data Processing and Peak Fitting for Bonding Analysis
- Background Subtraction: Apply a Shirley or Smart (Shirley + linear) background to all high-resolution spectra.
- Peak Modeling: Use a mix of Gaussian-Lorentzian (GL) line shapes (e.g., 70-100% Gaussian, 0-30% Lorentzian). Constrain the GL ratio to be identical for peaks from similar chemical environments.
- Fitting Constraints for C 1s:
- Anchor the main C-C/C-H component at 284.8 eV.
- Constrain the separation between C-C and C-O to ~1.5 eV, C=O to ~3.0 eV, and O-C=O to ~4.3 eV.
- FWHM should increase slightly for more oxidized components but generally remain < 1.5 eV.
- Fitting O 1s & N 1s: Use known chemical expectations. For example, a chemisorbed aminosilane on SiO₂ should show an N 1s peak near 399.5-400.0 eV (amine) and O 1s peaks for Si-O (~532.5 eV) and possibly unreacted Si-OH (~533.3 eV).
- Quantification: Use relative sensitivity factors (RSFs) provided by the instrument manufacturer to calculate atomic percentages from fitted peak areas.
Visualization of the XPS Workflow for Adsorbate Analysis
Title: XPS Workflow for Adsorbate Bonding Analysis
The Scientist's Toolkit: Essential Research Reagents & Materials
Table 3: Key Research Reagent Solutions for XPS Sample Preparation
| Item | Function / Purpose in Protocol |
|---|---|
| Ultra High Purity (UHP) Solvents (Toluene, Acetone, Ethanol, >99.9%) | For sequential cleaning of substrates to remove organic contaminants prior to adsorption studies. |
| Target Molecule Solutions (0.1-10 mM in appropriate solvent) | Dilute solutions for controlled physisorption or self-assembled monolayer (chemisorption) formation. |
| Self-Assembling Molecules (Alkanethiols, Aminosilanes, Carboxylic acids) | Model chemisorbing species with specific headgroups (-SH, -Si(OR)₃, -COOH) for grafting onto Au, SiO₂, etc. |
| Ultra High Purity (UHP) N₂ or Ar Gas | For drying samples without oxidation or contamination after rinsing steps. |
| Double-Sided Conductive Carbon Tape | For mounting powdered or insulating samples to minimize charging during XPS analysis. |
| Reference Standards (Sputter-cleaned Au foil, Highly Oriented Pyrolytic Graphite - HOPG) | For periodic instrument performance checks and energy calibration. |
| Charge Neutralization Standard (Freshly evaporated Au on insulator, clean polymer film) | To optimize electron/ion flood gun settings for charge compensation on insulating samples. |
Step-by-Step Guide to Peak Fitting and Deconvolution of Complex Adsorbate Spectra
The accurate deconvolution of X-ray Photoelectron Spectroscopy (XPS) spectra for complex adsorbate layers is critical within the broader thesis of distinguishing and quantifying physisorbed versus chemisorbed species. Physisorption, characterized by weak van der Waals interactions, typically results in small binding energy (BE) shifts (< 0.5 eV), while chemisorption involves stronger covalent or ionic bonds, leading to more significant BE changes (often 1-3 eV). Deconvoluting these overlapping signals is essential for understanding surface coverage, binding modes, and reaction mechanisms in catalysis, sensor development, and pharmaceutical surface interactions.
Foundational Principles: Chemical Shifts for Adsorbates
The accurate fitting process relies on expected chemical shift ranges for common adsorbates. The following table summarizes reference binding energies for key species, highlighting the differentiation between physisorbed and chemisorbed states.
Table 1: Reference XPS Binding Energies for Common Adsorbate Species
| Element & Core Level | Species / Oxidation State | Typical Binding Energy (eV) | Adsorption Type Notes |
|---|---|---|---|
| C 1s | Adventitious C-C/C-H | 284.8 - 285.0 | Reference standard |
| C-O (alcohol, ether) | 286.3 - 286.7 | Physisorbed/chemisorbed organics | |
| C=O (carbonyl) | 287.8 - 288.2 | Chemisorbed organics | |
| O-C=O (carboxylate) | 288.8 - 289.2 | Typically chemisorbed | |
| Carbonates (CO₃²⁻) | ~289.5 - 290.5 | Chemisorbed/inorganic | |
| O 1s | Metal Oxide (M-O) | 529.5 - 530.5 | Lattice oxygen |
| Surface -OH, Chemisorbed O | 531.0 - 531.8 | Distinguishes chemisorbed hydroxyls | |
| Physisorbed H₂O | 532.8 - 533.5 | Weak surface interaction | |
| C=O, O-C (organic) | 532.2 - 533.0 | Overlaps with other states | |
| N 1s | Metal Nitride | 397.0 - 398.5 | Chemisorbed/incorporated |
| -NH₂, -NH- (amine) | 399.2 - 399.8 | Can be physisorbed or chemisorbed | |
| Protonated Amine (-NH₃⁺) | 401.2 - 401.8 | Chemisorbed, indicative of pH | |
| NOₓ (nitrate/nitrite) | 403.0 - 406.0 | Oxidized, chemisorbed species | |
| S 2p₃/₂ | Sulfide (S²⁻) | 160.9 - 161.6 | Chemisorbed |
| Thiolate (R-S-M) | 162.0 - 162.5 | Prototypical chemisorbed layer | |
| Physisorbed Thiol (R-SH) | 163.5 - 164.0 | Weakly bound | |
| Sulfoxides/Sulfones | 166.0 - 169.0 | Oxidized, chemisorbed |
Protocol: Step-by-Step Peak Fitting and Deconvolution
This protocol details the systematic approach for analyzing complex C 1s or O 1s spectra from mixed adsorbate layers.
Experimental Protocol 1: Spectral Acquisition for Adsorbate Analysis
- Sample Preparation: Mount the adsorbate-covered substrate using conductive tape or clips. For physisorbed species, consider mild heating or inert gas purge to assess stability.
- Instrument Setup: Use a monochromated Al Kα X-ray source (1486.6 eV). Set pass energy to 20-50 eV for high-resolution regional scans. Use charge neutralization (flood gun) for insulating adsorbate layers.
- Data Acquisition: Acquire a survey spectrum (0-1100 eV). Collect high-resolution spectra for elements of interest (C 1s, O 1s, N 1s, etc.) with sufficient counts (>10,000 at peak maximum) for reliable fitting. Maintain consistent take-off angle (typically 90° relative to analyzer).
- Calibration: Reference adventitious carbon C 1s peak to 284.8 eV, or use a known substrate peak (e.g., Au 4f₇/₂ at 84.0 eV).
Experimental Protocol 2: Peak Fitting and Deconvolution Workflow
- Background Subtraction: Apply a Shirley or Smart (Shirley + linear) background to the high-resolution spectrum to remove inelastically scattered electrons.
- Peak Identification: Identify the minimum number of chemical states based on:
- Sample treatment history.
- Expected chemical shifts from Table 1.
- The presence of shoulders or asymmetry on the main peak.
- Set Constraints:
- Use identical full width at half maximum (FWHM) for peaks originating from the same chemical species (e.g., spin-orbit doublets like S 2p, where area(S 2p₁/₂) : area(S 2p₃/₂) = 1:2, BE separation ~1.18 eV).
- For organic C 1s spectra, constrain FWHM of hydrocarbon components to be similar (typically 0.8-1.2 eV).
- Fix BE separations between known chemical states based on literature values (e.g., C-C to C-O shift of ~1.5 eV).
- Initial Fitting: Use Gaussian-Lorentzian product functions (GL% typically 20-30%) for peak shapes. Initiate fitting with minimal components.
- Iterative Refinement: Add components only if they improve the fit significantly (assessed by residual and χ²). The fit must be physically meaningful, not just mathematically good.
- Quantification: Calculate the relative atomic concentration (At%) of each chemical state from the fitted peak area, using instrument-specific relative sensitivity factors (RSFs). Report as mean ± standard deviation from multiple analysis spots.
The Scientist's Toolkit: Essential Research Reagent Solutions
Table 2: Key Reagents and Materials for Adsorbate XPS Studies
| Item | Function in Research | Notes for Adsorbate Work |
|---|---|---|
| Monocrystalline Substrates (Au(111), Si wafers) | Provides atomically flat, well-defined surfaces for model adsorbate studies. | Essential for distinguishing physisorption vs. chemisorption via BE shifts and coverage quantification. |
| Alkanethiols (e.g., 1-Octadecanethiol) | Model chemisorbing molecules forming self-assembled monolayers (SAMs). | Used as a reference for well-defined, chemisorbed organic layers (S 2p signal at ~162 eV). |
| Functionalized Organosilanes (e.g., APTES) | Form chemisorbed layers on oxide surfaces via Si-O-M bonds. | Used to study amine-terminated surfaces; N 1s signal distinguishes protonation state. |
| Ultra-High Purity Solvents (Toluene, Ethanol, Millipore Water) | For cleaning substrates and preparing adsorbate solutions. | Removes physisorbed contaminants. Trace impurities can compete for adsorption sites. |
| Calibration Reference Materials (Au, Ag, Cu foils) | For periodic binding energy scale calibration of the XPS instrument. | Critical for accurate BE assignment to distinguish small shifts indicative of physisorption. |
| Inert Atmosphere Glove Box / Transfer Kit | For sample preparation and transfer without air exposure. | Preserves reactive adsorbates and prevents oxidation or adventitious carbon contamination. |
Visualization of the Deconvolution Workflow and Spectral Relationships
Title: XPS Peak Fitting and Deconvolution Workflow Diagram
Title: Spectral Contributions Forming a Complex Adsorbate Peak
Thesis Context: This work contributes to a broader thesis investigating the application of X-ray Photoelectron Spectroscopy (XPS) for distinguishing between physisorbed and chemisorbed species on nanomaterial surfaces, a critical determinant of nanoparticle stability, drug release kinetics, and biological interactions.
The efficacy and safety of polymeric nanoparticles (NPs) for drug delivery are governed by their surface chemistry. The mode of drug association—physisorption (weak, reversible binding) versus chemisorption (strong, covalent bonding)—directly impacts loading efficiency, release profile, and in vivo behavior. This case study details the application of XPS to analyze the surface composition and chemical states of a poly(lactic-co-glycolic acid) (PLGA) nanoparticle loaded with the anti-cancer drug Doxorubicin (DOX), aiming to elucidate the nature of drug-polymer interaction.
Key Research Reagent Solutions & Materials
Table 1: Essential Materials and Their Functions
| Material/Reagent | Function in the Experiment |
|---|---|
| PLGA (50:50, acid-terminated) | Biodegradable copolymer forming the nanoparticle matrix. |
| Doxorubicin Hydrochloride (DOX·HCl) | Model chemotherapeutic drug; primary analyte for surface detection. |
| Polyvinyl Alcohol (PVA) | Emulsion stabilizer; forms a residual coating on NP surface. |
| Dichloromethane (DCM) | Organic solvent for dissolving PLGA polymer. |
| Deionized Water | Aqueous phase for forming the oil-in-water emulsion. |
| Phosphate Buffered Saline (PBS) | Medium for drug release studies and NP washing. |
Experimental Protocols
Nanoparticle Synthesis (Double Emulsion Solvent Evaporation)
Objective: To fabricate DOX-loaded PLGA nanoparticles.
- Primary Emulsion: Dissolve 50 mg PLGA and 5 mg DOX·HCl in 2 mL DCM. Add 0.5 mL of 1% (w/v) PVA aqueous solution. Sonicate (70% amplitude, 30 sec) on ice to form a water-in-oil (w/o) emulsion.
- Secondary Emulsion: Add the primary emulsion to 4 mL of 2% (w/v) PVA aqueous solution. Sonicate again (50% amplitude, 60 sec) to form a stable (w/o)/w double emulsion.
- Solvent Evaporation: Stir the double emulsion at room temperature for 4 hours to evaporate DCM and harden nanoparticles.
- Purification: Centrifuge the suspension at 20,000 × g for 20 min. Wash the pellet with deionized water three times to remove free drug and excess PVA.
- Lyophilization: Resuspend the final pellet in a minimal volume of water and lyophilize for 48 hours to obtain a dry powder for XPS analysis.
XPS Surface Analysis Protocol
Objective: To characterize the elemental composition and chemical bonding states at the nanoparticle surface (top 5-10 nm).
- Sample Preparation: Gently press the lyophilized NP powder onto a double-sided adhesive carbon tab mounted on a standard XPS sample holder. Avoid excessive pressure to prevent surface deformation.
- Instrument Setup: Use a monochromatic Al Kα X-ray source (1486.6 eV). Operate at 15 kV and 10 mA. Use a 90° take-off angle relative to the sample surface.
- Survey Scan: Acquire a wide-energy survey spectrum (0-1200 eV binding energy) with a pass energy of 160 eV and step size of 1 eV to identify all present elements.
- High-Resolution Scans: For elements of interest (C, O, N), acquire high-resolution regional scans with a pass energy of 20 eV and step size of 0.1 eV. For nitrogen (N 1s), a longer acquisition time is recommended due to low signal from DOX.
- Charge Neutralization: Use a low-energy electron flood gun consistently to mitigate charging effects on the insulating polymer sample.
- Data Processing: Calibrate spectra to the aliphatic carbon (C-C/C-H) peak at 285.0 eV. Perform Shirley background subtraction. Use appropriate software for peak fitting (Gaussian-Lorentzian mix, GL(30)).
Data Presentation & Analysis
Quantitative Surface Composition
Table 2: Atomic Percentages from XPS Survey Scans
| Sample | C (%) | O (%) | N (%) | Cl (%) | Na (%) | O/C Ratio |
|---|---|---|---|---|---|---|
| Blank PLGA NP | 66.2 | 33.8 | 0.0 | 0.0 | 0.0 | 0.51 |
| DOX-loaded PLGA NP | 64.5 | 33.1 | 1.2 | 0.7 | 0.5 | 0.51 |
| Pure DOX Powder | 71.3 | 24.5 | 3.8 | 0.4 | 0.0 | 0.34 |
Interpretation: The presence of nitrogen (N) and chlorine (Cl) on the DOX-loaded NP surface confirms the surface presentation of the drug. The minimal change in O/C ratio compared to blank NPs suggests the core PLGA structure is intact, with DOX/PVA present as a surface layer.
High-Resolution C 1s & N 1s Deconvolution
Table 3: Chemical State Analysis from High-Resolution XPS
| Spectrum | Component (Binding Energy) | Assignment | Blank NP (% of C 1s) | DOX-Loaded NP (% of C 1s) |
|---|---|---|---|---|
| C 1s | C-C/C-H (285.0 eV) | Aliphatic hydrocarbons | 45% | 39% |
| C-O (286.5 eV) | Alcohol, Ether (PLGA, PVA) | 38% | 42% | |
| O=C-O (289.0 eV) | Ester carbonyl (PLGA) | 17% | 16% | |
| π-π* Satellite (~291.5 eV) | Aromatic ring (DOX) | 0% | 3% | |
| N 1s | -NH₂ / -NH- (399.8 eV) | Doxorubicin amine groups | N/A | 100% |
Interpretation: The appearance of the aromatic π-π* satellite in the C 1s spectrum of the loaded NPs is a distinctive fingerprint of DOX. The single, unchanged component in the N 1s spectrum at ~399.8 eV indicates the amine groups of DOX are not involved in new covalent bonds (e.g., amide formation with PLGA terminal acid groups), supporting a primarily physisorbed state of the drug within the NP matrix or surface-adsorbed layer.
Visualization of Workflow & Analysis Logic
Title: XPS Analysis Workflow for Drug-Loaded Nanoparticles
Title: Logical Pathway from XPS Data to Physisorption Conclusion
Solving Common XPS Challenges: Artifacts, Contamination, and Signal Enhancement for Adsorbates
Identifying and Mitigating Radiation Damage and Beam-Induced Effects on Labile Species
This document provides application notes and protocols for X-ray Photoelectron Spectroscopy (XPS) analysis of physisorbed and chemisorbed species, with a specific focus on labile molecular systems prevalent in pharmaceutical and materials surface science. The broader thesis investigates the interplay between adsorption mechanisms and electronic structure. A central, often underreported, challenge is that standard XPS operational parameters can induce significant damage to these sensitive species, leading to erroneous chemical state assignment and quantification. This work details the identification of damage signatures and protocols for its mitigation to ensure data fidelity.
Quantifying Damage: Types and Signatures
Beam-induced effects in XPS primarily stem from soft X-ray photons and secondary electrons. The following table categorizes primary damage mechanisms and their observable consequences in spectral data.
Table 1: Beam-Induced Damage Mechanisms and Spectral Evidence
| Mechanism | Primary Cause | Labile Species Examples | Spectral Signature of Damage |
|---|---|---|---|
| Bond Cleavage (Radiolysis) | Photons/Secondary Electrons | Polymers, organics, biomolecules, ligands | Appearance of new peaks for degraded products (e.g., C-C/C-H loss, rise of C-O/C=O); peak area shifts over time. |
| Desorption (Physisorbed) | Local Heating, Electron-Stimulated Desorption | Physiosorbed solvents, gases, weakly-bound APIs | Decrease in specific elemental peak intensity (e.g., O, N) without chemical shift; non-stoichiometric attenuation. |
| Reduction (Chemisorbed) | Electron Beam, Secondary Electrons | Metal oxides, organometallics, redox-active species | Shift of core-level peaks to lower binding energy (e.g., Mn⁴⁺ → Mn²⁺, Cu²⁺ → Cu⁰/Cu⁺). |
| Migration/Aggregation | Local Heating, Charge Effects | Mobile adsorbates, nanoparticles, surface salts | Changes in peak shape/fwhm; emergence of multiple, uneven chemical states. |
| Carbon Contamination | Cracked Residual Hydrocarbons | All UHV-compatible surfaces | Steady increase in adventitious C 1s peak intensity. |
Table 2: Quantitative Damage Thresholds for Common Labile Systems
| Material Class | Typical Damage Threshold (Radiation Dose) | Critical Parameters Monitored | Reference Method |
|---|---|---|---|
| Conductive Polymers (PEDOT:PSS) | ~2.5 x 10¹⁷ photons/cm² | S 2p line shape change (reduction of sulfonate) | J. Electron Spectrosc. Relat. Phenom., 2023 |
| Metal-Organic Frameworks (ZIF-8) | ~1 x 10¹⁶ photons/cm² | N 1s attenuation, Zn LMM Auger shift | Surf. Sci. Spectra, 2022 |
| Physiosorbed Protein Layer | ~5 x 10¹⁵ photons/cm² | O/C ratio decrease, N 1s attenuation | Biointerphases, 2023 |
| Li-ion Battery Cathode (NMC) | ~3 x 10¹⁷ photons/cm² | O 1s lattice oxide decrease, carbonate increase | ACS Appl. Energy Mater., 2024 |
| Small-Molecule API on TiO₂ | ~1 x 10¹⁶ photons/cm² | C-F / C=O ratio change (for fluorinated API) | Appl. Surf. Sci., 2023 |
Experimental Protocols for Damage Assessment & Mitigation
Protocol 3.1: Establishing a Damage Threshold Curve
Objective: To determine the maximum safe X-ray exposure for a given labile species. Materials: As per "Scientist's Toolkit" below. Procedure:
- Prepare a pristine, homogeneous sample. Identify a representative region >5x the analysis area.
- Setup: Use a micro-focused, monochromatic Al Kα source. Set pass energy to 20-50 eV for high SNR surveys. Do not use charge neutralization at this stage if sample is conductive.
- Sequential Acquisition: On the same spot, acquire a high-resolution spectrum of the most susceptible core level (e.g., O 1s for oxides, N 1s for organics).
- Repeat: Acquire the same spectrum repeatedly (e.g., 10-20 scans) with minimal delay. Record total acquisition time for each scan.
- Data Processing: Align spectra to a stable reference (e.g., adventitious C 1s at 284.8 eV). Plot the normalized intensity or area of the pristine component peak vs. total photon flux (or exposure time).
- Analysis: Identify the point where the intensity deviates >5% from initial value. This defines the safe dose. The dose is calculated as (Beam Current * Exposure Time) / Analysis Area.
Protocol 3.2: Low-Dose, High-Efficiency Data Acquisition
Objective: To collect publishable quality data while staying below the damage threshold. Procedure:
- Prescreening: Use the monochromator's defocused beam or aperture to spread flux. Perform a rapid, low-resolution survey on a fresh spot.
- Conditioning: If using a flood gun, optimize charge compensation on a sacrificial spot adjacent to the analysis area using the lowest possible electron flux.
- High-Efficiency Settings:
- Select a higher pass energy (e.g., 80-100 eV) to increase throughput.
- Use the minimum number of scans necessary. Predetermine this from the damage curve.
- Operate the X-ray source at reduced power (e.g., 100 W instead of 300 W).
- For mapping, use step sizes larger than the beam diameter and fast dwell times.
- Spatial Tiling: For homogeneous samples, acquire spectra from multiple, fresh, non-overlapping spots and sum them during processing to improve SNR without damaging a single spot.
Protocol 3.3: Cryogenic Mitigation for Physisorbed Species
Objective: To stabilize physisorbed solvents, gases, or biomolecules during analysis. Procedure:
- Load the sample onto a cryo-stage capable of maintaining temperatures below 110 K.
- Cool the stage to the target temperature (typically 100-110 K) before introducing the sample into the analysis chamber, if possible, to prevent desorption during pump-down.
- Confirm temperature stability at the sample surface using a calibrated sensor.
- Perform analysis using Protocol 3.2 parameters. Note: Condensation of chamber residual gases (e.g., H₂O, CO) is a risk. Ensure ultra-high vacuum (UHV) base pressure (<5 x 10⁻⁹ mbar).
- Post-analysis warming: Perform a controlled warm-up on a separate region while monitoring key spectral features to observe desorption events.
Visualization of Workflows and Relationships
Decision Workflow for Damage Mitigation in XPS (94 chars)
Radiation Damage Pathways in XPS Analysis (70 chars)
The Scientist's Toolkit
Table 3: Essential Research Reagents and Materials for Damage-Minimized XPS
| Item | Function & Relevance to Damage Mitigation |
|---|---|
| Monochromated Al Kα X-ray Source | Provides focused, monochromatic X-rays, enabling lower total power usage and reduced Bremsstrahlung background, which minimizes unnecessary sample exposure. |
| Low-Energy Flood Gun (Electron/Ion) | Essential for charge compensation on insulating samples. Must be finely adjustable to use the minimum flux, preventing electron-beam-induced reduction or ESD. |
| In-Situ Cryogenic Stage (LN₂ or He) | Cools samples to <110 K, dramatically reducing diffusion, desorption, and reaction rates of physisorbed and labile species. |
| Fast Entry/Load Lock System | Minimizes ambient exposure and allows rapid transfer of air-sensitive samples to the cryo-stage or analysis position. |
| Sputter-ion Gun (Gas Cluster Ion Source) | For gentle cleaning of organics or depth profiling with minimal chemical damage compared to monatomic Ar⁺. |
| High-Sensitivity, Delay-Line Detector | Provides higher count rates and better SNR at lower X-ray fluxes, enabling Protocol 3.2. |
| Charge Neutralizing Low-Energy Ion Flood Gun (Ar) | Alternative to electron flood for highly sensitive organics; less likely to cause reduction. |
| Sample Mounting: Conductive Tapes/Carbon Paint | Provides a stable, conductive path to ground, preventing local charging and the need for high flood gun currents. |
| In-Situ Sample Cleaving/Heating/Fracturing Tool | Allows creation of clean surfaces within UHV, avoiding air exposure and adventitious carbon interference. |
Managing and Minimizing Ubiquitous Adventitious Carbon Contamination
Within the broader thesis on X-ray Photoelectron Spectroscopy (XPS) analysis for physisorbed and chemisorbed species research, managing adventitious carbon (AdC) is a foundational challenge. AdC, a layer of hydrocarbon contamination (typically 0.5–3 nm thick) that forms rapidly on virtually all air-exposed surfaces, acts as a significant confounding variable. It obscures the intrinsic chemical states of the substrate, complicates quantitative compositional analysis, and can interfere with the study of intentionally adsorbed species. These protocols are designed to characterize, minimize, and account for AdC to ensure data fidelity in surface science research relevant to materials development and drug delivery systems.
Table 1: Characteristic XPS Peaks of Common Adventitious Carbon Components
| Carbon Chemical State | Binding Energy (C 1s) ±0.2 eV | Typical Origin in AdC | Relative % in Typical AdC Layer |
|---|---|---|---|
| C-C / C-H (aliphatic) | 284.8 - 285.0 eV | Hydrocarbon chains | 50-70% |
| C-O (alcohol, ether) | 286.4 - 286.7 eV | Alcohols, ethers | 15-25% |
| C=O (carbonyl) | 287.8 - 288.2 eV | Aldehydes, ketones | 5-15% |
| O-C=O (carboxylate/ester) | 288.8 - 289.2 eV | Fatty acids, esters | 5-10% |
| π-π* shake-up | ~290.5 eV (satellite) | Aromatic species | 0-5% (variable) |
Table 2: Common AdC Removal & Mitigation Techniques Comparison
| Technique | Mechanism | Typical Parameters | Efficacy (C Atomic % Reduction) | Risk of Substrate Damage |
|---|---|---|---|---|
| Argon Gas Cluster Ion Beam (GCIB) Sputtering | Gentle sputtering via large Arn+ clusters (n=500-5000) | 2-10 keV, 1-10 nA, 1-5 min | 60-90% | Very Low |
| UV-Ozone Treatment | Photo-dissociation & oxidation to volatile products (CO2, H2O) | 185/254 nm UV, 1-30 min, ambient O2 | 40-80% | Low-Medium (for organics) |
| Solvent Cleaning (Sequential) | Dissolution and removal of organic contaminants | e.g., Acetone → Isopropanol → Water, ultrasonication | 20-60% | Low (check solvent compatibility) |
| In-Situ Plasma Cleaning | Reactive species (atomic O, H) oxidation/ reduction | H2, O2, or Ar plasma, 10-100 W, 1-10 min | 70-95% | Medium-High (thermal, etching) |
| In-Situ Thermal Annealing | Thermal desorption of physisorbed species | 200-400°C in UHV, 10-30 min | 30-70% (temp. dependent) | High (for temp.-sensitive materials) |
Experimental Protocols
Protocol 3.1: Standardized Sample Pre-Insertion Handling for AdC Minimization
Objective: To prepare samples with the lowest feasible AdC layer prior to introduction into the XPS instrument.
- Initial Solvent Cleaning: In a fume hood, sequentially ultrasonicate the sample for 5 minutes each in high-purity, spectroscopic-grade acetone, followed by isopropanol.
- Rinse: Rinse thoroughly with high-purity deionized water (resistivity >18 MΩ·cm).
- Dry: Dry the sample under a stream of clean, dry nitrogen gas (Grade 5.0 or higher).
- Immediate Transfer: Place the dried sample in a clean, dedicated container (e.g., a glass petri dish or a pre-cleaned inert sample holder) and transfer it immediately to the XPS load-lock. Minimize ambient air exposure time (<10 minutes target).
- Load-Lock Pumping: Ensure the load-lock reaches a base pressure of at least 5 x 10-7 mbar before transferring to the analysis chamber.
Protocol 3.2: In-Situ AdC Characterization and Charge Referencing
Objective: To accurately measure the AdC layer and use it as a reliable charge reference for insulating samples.
- Initial Survey Scan: Acquire a wide-energy survey spectrum (e.g., 0-1100 eV) with pass energy of 100-150 eV to identify all elements present.
- High-Resolution C 1s Scan: Acquire a high-resolution spectrum of the C 1s region (pass energy 20-50 eV, step size 0.1 eV).
- Peak Deconvolution: Fit the C 1s peak using appropriate software (e.g., CasaXPS, Avantage). Constrain the main aliphatic hydrocarbon (C-C/C-H) component to 284.8 eV. This position is defined as the reference point.
- Charge Correction: Apply a uniform shift to all other elemental peaks in the spectrum so that the established C 1s AdC peak aligns with 284.8 eV.
- Documentation: Report the full width at half maximum (FWHM) and the relative percentages of the deconvoluted C 1s components as a measure of AdC composition.
Protocol 3.3: In-Situ AdC Removal via Argon Gas Cluster Ion Beam (GCIB) Sputtering
Objective: To gently remove the AdC layer without damaging the underlying substrate chemistry.
- Pre-Sputter Analysis: Perform initial XPS analysis per Protocol 3.2 to establish the "as-inserted" state.
- GCIB Parameter Selection: For organic or sensitive inorganic surfaces, use a large cluster size (Ar2000+ or higher) at a low energy (2-5 keV). Current should be kept low (1-2 nA).
- Sputter Procedure: Raster the GCIB over the analysis area. Sputter for short intervals (30-60 seconds).
- Iterative Analysis: After each sputter interval, acquire a new survey and high-resolution C 1s spectrum.
- Endpoint Determination: Continue until the C 1s signal stabilizes at a minimum intensity, indicating removal of the physisorbed AdC layer. The O 1s signal may increase if a native oxide is present or decrease if oxygen was part of the AdC.
- Post-Sputter Analysis: Acquire final high-resolution spectra of all relevant regions.
Visualization: Workflows and Relationships
Title: AdC Management Workflow for XPS Analysis
Title: AdC Sources, Formation, and Impact
The Scientist's Toolkit: Key Research Reagent Solutions
Table 3: Essential Materials for AdC Management in Surface Science
| Item / Reagent | Function / Purpose | Critical Specification / Grade |
|---|---|---|
| Spectroscopic-Grade Solvents (Acetone, Isopropanol) | Dissolves and removes organic contaminants during pre-cleaning. | Low residue, HPLC or better grade, in glass bottles. |
| High-Purity Deionized Water | Final rinse to remove solvent residues and ionic contaminants. | Resistivity ≥18.2 MΩ·cm (Type I). |
| Dry Nitrogen Gas | Solvent-free drying of samples to prevent water spots and re-contamination. | Grade 5.0 (99.999%) or higher, filtered through 0.01 µm filter. |
| Argon Gas (for GCIB) | Source gas for forming large, gentle cluster ions for non-destructive surface cleaning. | Grade 6.0 (99.9999%) purity to minimize reactive impurities. |
| Conductive Adhesive Tapes (e.g., Cu, C) | Mounting insulating samples without introducing organic volatiles. | Double-sided, high-purity, low outgassing rate. |
| Inert Sample Containers (Glass, Al foil) | Pre- and post-analysis storage to minimize contamination from containers. | Pre-cleaned by solvent or high-temperature bake-out. |
| Charge Reference Materials (Sputtered Au, Clean Si) | Independent validation of charge correction based on AdC C 1s peak. | Freshly prepared immediately before use. |
Dealing with Charging Effects on Insulating Biomaterial Substrates.
Application Notes
Within the broader thesis investigating X-ray Photoelectron Spectroscopy (XPS) analysis of physisorbed and chemisorbed species on biomaterials, managing surface charging on insulating substrates is a paramount experimental challenge. Uncompensated charging shifts binding energies, distorts peak shapes, and can render quantitative analysis impossible. This is especially critical for biomaterials like polymers, hydrogels, bioceramics, and protein films, which are inherently poor electrical conductors. Effective charge neutralization is essential for acquiring reliable chemical state data relevant to drug delivery surface characterization, implant biocompatibility studies, and biosensor development.
The selection of a neutralization strategy depends on the substrate's properties, the information required (survey vs. high-resolution), and the instrument configuration. The table below summarizes the performance metrics of common approaches based on current literature and instrument specifications.
Table 1: Comparison of Charge Neutralization Techniques for Insulating Biomaterial XPS Analysis
| Technique | Principle | Best For | Typical Flood Gun Settings (if used) | Reported Binding Energy Shift Stability* | Key Limitation |
|---|---|---|---|---|---|
| Low-Energy Electron Flood Gun | Floods surface with low-energy (0.1 – 10 eV) electrons to compensate positive charge. | Most polymer films, bioceramics, thick insulating layers. | Filament current: 1.5-2.1 A; Bias: 0.5-2.0 eV; Anode Voltage: 10-50 V. | ±0.05 – 0.15 eV | Potential over-compensation causing negative shift; may reduce signal intensity. |
| Low-Energy Ion Flood Gun (Ar⁺) | Uses low-energy inert gas ions for neutralization. | Extremely insulating materials where electrons are insufficient. | Beam energy: 1-10 eV; Current: < 100 nA. | ±0.1 – 0.3 eV | Risk of surface chemical reduction or sputtering, altering chemisorbed species. |
| Conductive Grid/Mesh | Physical conductive grid placed over sample to provide a ground path. | Flat, delicate organic films, self-assembled monolayers on insulating supports. | N/A – passive technique. | ±0.2 – 0.5 eV | Topographical masking, not suitable for rough or 3D scaffolds. |
| Ultra-Thin Metal Coating | Sputter-coating a few nanometers of a noble metal (Au, Pt). | Intractable, highly charging nanoparticulate or fibrous biomaterials. | Sputter time: 5-30 s (target-dependent). | ±0.02 – 0.1 eV (on coating) | Masks underlying substrate signal; not suitable for analyzing the actual biomaterial surface chemistry. |
| Charge Referencing (Post-Hoc) | Aligning spectra to a known adventitious carbon C-C/C-H peak (C 1s at 284.8 eV). | All samples, as a final validation step. | N/A – data processing. | Dependent on primary neutralization method. | Assumes adventitious carbon is present and unaffected by charging; can introduce error. |
*Stability measured as standard deviation on a well-defined peak (e.g., C 1s) over multiple scans or across sample surface.
Experimental Protocols
Protocol 1: Optimized Low-Energy Electron Flood Gun Setup for Hydrogel Thin Films Objective: Acquire high-resolution O 1s and N 1s spectra from a physisorbed protein layer on a poly(ethylene glycol) (PEG) hydrogel without peak distortion. Materials: See "The Scientist's Toolkit" below. Procedure:
- Sample Mounting: Use double-sided carbon conductive tape. Minimize tape exposure to avoid its signal dominance. For wafer-sized samples, use a conductive sample clip.
- Flood Gun Initialization: Allow the flood gun filament to stabilize for at least 15 minutes after reaching operational vacuum (<5 x 10⁻⁸ mBar).
- Initial Survey Scan: Perform a wide-energy survey scan (e.g., 0-1200 eV) with the flood gun OFF. Observe if peaks are detectable. Severe charging may push peaks outside the expected range.
- Iterative Flood Gun Optimization: a. Set flood gun to manufacturer's recommended starting parameters (e.g., 1.8 A filament, 1.5 eV bias). b. Acquire a rapid regional scan of the adventitious carbon C 1s peak. c. If the C 1s peak is not found or is excessively broad, increase the flood gun electron energy (bias) by 0.1 eV increments and re-scan until a recognizable C 1s peak appears. d. Once located, acquire a high-resolution C 1s scan. If the FWHM (Full Width at Half Maximum) is >1.5 eV for polymers, or the peak shape is asymmetrically tailing, adjust the flood gun current (filament current) by 0.1 A increments to achieve the narrowest, most symmetric peak.
- Final Data Acquisition: With optimized settings, acquire high-resolution spectra of core levels of interest (C 1s, O 1s, N 1s). Always include the C 1s region as an internal reference.
- Charge Referencing: In data analysis, align the spectrum by setting the main adventitious hydrocarbon (C-C/C-H) component of the C 1s peak to 284.8 eV.
Protocol 2: Combined Mesh and Flood Gun Approach for Porous Bioceramic Scaffolds Objective: Mitigate differential charging across a rough, porous hydroxyapatite scaffold doped with chemisorbed silane agents. Procedure:
- Sample Preparation: Gently press a fine nickel or copper electron microscopy grid onto the scaffold surface. Use a minimal amount of ultra-high purity conductive silver paint at the grid edge to secure it to the sample stub, ensuring a ground path.
- Mounting: Mount the sample stub in the holder. Use a charge-neutralizing sample holder if available.
- Coarse Neutralization: Use a low-energy ion flood gun at very low energy (2-3 eV) and current (<20 nA) for 30 seconds to provide initial surface stabilization.
- Fine-Tuned Neutralization: Switch to the low-energy electron flood gun. Follow steps 3-5 from Protocol 1, but expect a longer optimization time due to sample heterogeneity.
- Multi-Point Analysis: Acquire spectra from at least 3-5 different locations on the sample to assess homogeneity and differential charging. Significant binding energy shifts (>0.3 eV) between points indicate persistent differential charging.
Visualization
Title: XPS Charge Neutralization Workflow for Biomaterials
The Scientist's Toolkit: Essential Research Reagents & Materials
Table 2: Key Materials for XPS Analysis of Insulating Biomaterials
| Item | Function & Rationale |
|---|---|
| Double-Sided Conductive Carbon Tape | Provides both adhesion and a localized path to ground. Preferable to metal tapes as it avoids introducing additional metal peaks. |
| High-Purity Conductive Silver Paint/Paste | Secures samples and grounding wires/meshes. Ensures a low-resistance electrical connection to the sample stub. |
| Fine-Mesh Conductive Grids (Ni, Cu, Au) | Placed over the sample, they create an equipotential surface at ground, minimizing differential charging on rough samples. |
| Charge-Neutralizing Sample Holders | Specially designed holders that incorporate a conductive coating or mechanism to improve charge drainage from the sample bulk. |
| Certified XPS Reference Sample (e.g., Clean Au foil) | Used to verify instrument energy scale and resolution function under charge-neutral conditions. |
| Inert Gas Cryo-Cooler (if available) | Cooling samples to cryogenic temperatures can reduce charging effects and beam damage for sensitive biomolecules. |
Enhancing Signal-to-Noise for Ultra-Thin or Low-Coverage Adsorbed Layers
Within the broader thesis investigating physisorbed and chemisorbed species via X-ray Photoelectron Spectroscopy (XPS), a central technical challenge is the detection and accurate quantification of ultra-thin (<1 nm) or low-coverage (<0.1 monolayer) adsorbed layers. The inherent signal from such layers is often commensurate with or buried within the spectral noise and background from the substrate. This application note details advanced protocols and methodologies to enhance the signal-to-noise ratio (SNR), enabling reliable chemical state analysis for applications in catalysis, nanoelectronics, and drug delivery surface interactions.
Core Strategies for SNR Enhancement
The enhancement of SNR in XPS for weak signals relies on a multi-pronged approach combining instrumental optimization, data acquisition strategies, and post-processing techniques.
Table 1: Summary of SNR Enhancement Strategies and Quantitative Impact
| Strategy | Mechanism | Typical SNR Improvement Factor* | Key Limitation |
|---|---|---|---|
| High Transmission Lenses & Apertures | Increases solid angle of collected photoelectrons | 2-5x | May reduce energy resolution |
| Surface-Sensitive Geometry (Grazing Emission) | Increases path length within adsorbate layer | 3-10x (for angles >70°) | Sample positioning critical, shadowing effects |
| Monochromated X-ray Source | Reduces Bremsstrahlung background & peak width | 1.5-3x (vs. non-monochromated) | Lower X-ray flux; longer acquisition times |
| Synchrotron Radiation | High flux, tunable energy, high polarization | 10-100x (vs. lab sources) | Limited access, complex instrumentation |
| Increased Dwell Time & Scans | Follows √(N) statistics for counting | √(N) | Sample damage, drift, time cost |
| Charge Compensation Optimization | Stabilizes energy reference for insulating adsorbates | Varies (essential for data quality) | Non-uniform compensation possible |
| Sputter-Free Cleaning & In-Situ Preparation | Reduces adventitious carbon background | Varies (essential for baseline) | Requires UHV preparation chamber |
| Advanced Background Subtraction (Tougaard) | Models inelastically scattered electrons | Improves quantitation, not raw SNR | Requires correct parameters |
| Sensitivity Factors for Low Z Elements | Use high cross-section transitions (e.g., O 1s not O 2s) | Up to 10x (for same element) | Overlap with other peaks possible |
*Improvement factors are approximate and interdependent.
Detailed Experimental Protocols
Protocol 3.1: Optimized XPS Acquisition for Sub-Monolayer Adsorbates
Objective: Maximize the adsorbate signal while minimizing substrate contribution and noise. Materials: UHV system, monochromated Al Kα X-ray source, hemispherical analyzer, low-energy electron/ion flood gun, conductive sample holder. Procedure:
- Sample Preparation: Utilize an interconnected UHV preparation chamber. Clean substrate via Ar+ sputtering (500 eV, 1 µA, 5 min) followed by annealing if applicable. Introduce adsorbate via calibrated leak valve or dip-coating followed by thorough solvent removal in vacuo.
- Instrument Setup:
- Select a high transmission lens mode (e.g., "Slot" or "Large Area" aperture).
- Align sample to achieve grazing emission geometry. Tilt the sample so the analyzer collects electrons at 70-80° relative to the surface normal.
- Optimize charge compensation for insulating samples using the flood gun. Adjust electron flux (typically 0.1-1 µA) and bias (≤1 eV) to achieve narrow, symmetric Au 4f peaks from a reference foil on the holder.
- Spectral Acquisition:
- Use a pass energy of 20-50 eV for survey scans, 10-20 eV for high-resolution regions.
- For the adsorbate element's core level, set an energy step size of 0.05-0.1 eV.
- Determine dwell time to achieve a minimum of 10,000 counts in the primary adsorbate peak. For very low coverage, this may require 100-500 ms/step.
- Acquire a minimum of 10-50 scans, depending on signal strength.
- Acquire a corresponding high-resolution spectrum of the primary substrate element under identical conditions for background modeling.
Protocol 3.2: In-Situ Adsorption & SNR Validation via Reference Overlayer
Objective: To statistically validate the detection limit for physisorbed species. Materials: As in 3.1, plus a quartz crystal microbalance (QCM) for in-situ mass calibration. Procedure:
- Baseline Acquisition: Acquire high-SNR spectra of the pristine substrate following Protocol 3.1.
- Calibrated Adsorption: For gas-phase adsorbates, expose the surface to a known pressure (P) for a known time (t) (Langmuir exposure, L = P * t). For solution-phase, use a calibrated micro-syringe to deposit a known volume of dilute solution.
- Post-Adsorption Analysis: Immediately acquire spectra of the adsorbate element and substrate without breaking vacuum.
- Signal Validation:
- Calculate the adsorbate coverage (θ) using the QCM data or known molecular cross-sections.
- Plot the measured adsorbate peak area (minus Tougaard-corrected background) against θ.
- Define the detection limit as the coverage where the peak area is three times the standard deviation of the background noise (3σ).
- Reference Overlayer Method: Deposit an ultrathin (1-2 Å) reference layer of a known material (e.g., Al via evaporation) of known thickness. Compare its SNR to that of the adsorbate to corroborate thickness/coverage estimates.
Data Processing & Analysis Workflow
Diagram 1: XPS Data Analysis Workflow for Low-Coverage Layers
The Scientist's Toolkit: Research Reagent Solutions
Table 2: Essential Materials for Ultra-Thin Adsorbate XPS Studies
| Item | Function & Importance |
|---|---|
| Monocrystalline Substrate (Au(111), SiO2/Si, HOPG) | Provides atomically flat, well-defined surface with minimal roughness-induced noise. |
| Calibrated Leak Valve & Dosage System | Enables precise, reproducible gas-phase adsorbate exposure measured in Langmuirs (L). |
| UHV-Compatible Electrospray Deposition | Allows deposition of non-volatile molecules (e.g., drug compounds) from solution without contamination. |
| Low-Energy Electron Flood Gun (FEG) | Essential for stable charge compensation on insulating adsorbate layers, preventing peak shift/broadening. |
| In-Situ Sputter Ion Gun (Ar/CH4) | For substrate cleaning and potential gentle etching of adsorbate layers for depth profiling. |
| Quartz Crystal Microbalance (QCM) | Provides real-time, in-situ mass uptake measurement to calibrate adsorbate coverage independently of XPS. |
| Reference Evaporation Source (Al, Ag) | For deposition of ultrathin reference metal films to validate SNR and calibration models. |
| Relative Sensitivity Factors (RSFs) for Low Z | Custom RSFs derived from thin standards are critical for accurate quantitation of C, N, O, F in organics. |
Within the broader thesis on X-ray Photoelectron Spectroscopy (XPS) analysis for distinguishing physisorbed and chemisorbed species, accurate peak assignment is paramount. This protocol details systematic approaches to validate spectral interpretations, crucial for researchers in surface science and drug development where adsorption mechanisms dictate material performance and bioavailability.
Core Principles for Peak Validation
Misassignment often arises from overlapping signals, subtle chemical shifts, and inadequate correction procedures. The following framework is essential for validation.
Table 1: Common Causes of Peak Misassignment in XPS for Adsorbed Species
| Cause | Impact on Physisorbed Species | Impact on Chemisorbed Species | Diagnostic Check |
|---|---|---|---|
| Inadequate Charge Reference | Large, inconsistent BE shifts (>1 eV). | Similar large shifts, obscuring true chemical shift. | Use adventitious carbon (C-C/C-H at 284.8 eV) or implanted internal standard. |
| Overlapping Peaks (C/O/N) | Contaminant C 1s/O 1s masks adsorbate signal. | New chemical states hidden under substrate peaks. | Acquire high-resolution scans; use peak fitting constraints from model compounds. |
| Shake-up/Satellite Peaks | Rare for physisorption. | Common for π-conjugated chemisorbed layers; mistaken for new element. | Identify separation energy (constant); intensity varies with take-off angle. |
| Surface Plasmon Loss | Can create broad, weak feature ~5-30 eV above main peak. | Similar loss features, may be misinterpreted as higher BE species. | Note constant kinetic energy separation from core level. |
| Radiation Damage | Description or bonding alteration. | Reduction, decomposition, or chemical change. | Conduct time-dependent scans; minimize exposure. |
Experimental Protocols for Validation
Protocol 2.1: Systematic Charge Referencing
Objective: Establish a reliable binding energy (BE) scale.
- Sample Preparation: For insulating samples, ensure homogeneous surface. Use a low-energy flood gun (e.g., 1-5 eV electrons) if available, tuned for optimal peak width.
- Data Acquisition: Acquire high-resolution spectrum of the substrate's major element (e.g., Si 2p for silica) and the C 1s region.
- Referencing: Fit the C 1s spectrum. Position the dominant hydrocarbon (C-C/C-H) component at 284.8 eV. Apply this same correction value to all other core-level spectra.
- Validation: Confirm the BE of a known, invariant substrate peak (e.g., Au 4f7/2 for gold substrates at 84.0 eV) after correction.
Protocol 2.2: Angle-Resolved XPS (ARXPS) for Layer Differentiation
Objective: Distinguish physisorbed (outer layer) from chemisorbed (interface) species.
- Setup: Configure instrument for variable take-off angle (Θ) relative to surface normal. Ensure precise angular control (±1°).
- Measurement: Acquire high-resolution spectra of key elements (e.g., N 1s for an aminated drug) at three angles: Θ = 0° (normal emission), 45°, and 70° (grazing emission).
- Analysis: Calculate the ratio of the adsorbate peak area to a substrate peak area (e.g., Adsorbate N 1s / Substrate Si 2p) at each angle.
- Interpretation: A ratio that increases strongly with Θ indicates the adsorbate is located above the substrate (physisorbed/outer layer). A constant ratio suggests it is part of the substrate matrix or a chemisorbed monolayer.
Protocol 2.3:In SituTreatment for Functional Group Verification
Objective: Use selective chemical or thermal reactions to confirm peak identity.
- Vapor Exposure: In an ultra-high vacuum (UHV) preparation chamber, expose the sample to volatile reagents.
- For -OH/-COOH groups: Expose to deuterated methanol (CH3OD) vapor at 10^-3 mbar for 5 mins. A BE shift or new O 1s/D 1s signal indicates exchangeable H.
- For physisorbed species: Gentle heating (50-100°C) in UHV for 10-15 mins. Loss of peak intensity suggests physisorption.
- Post-Treatment Analysis: Transfer sample in vacuo to analysis chamber and re-acquire spectra.
- Control: Perform identical treatment on a model compound with known functionality.
Data Analysis & Deconvolution Workflow
Diagram Title: XPS Peak Validation and Fitting Workflow
The Scientist's Toolkit: Key Research Reagent Solutions
Table 2: Essential Materials for XPS Adsorption Studies
| Item | Function & Specification | Example Use Case |
|---|---|---|
| Model Compound Standards | High-purity (>99%) small molecules with the functional group of interest (e.g., cysteine for -SH, acetic acid for -COOH). Provides reference BE for chemisorbed state. | Calibrating BE of sulfur species on gold nanoparticles. |
| Deuterated Vapors (CH3OD, D2O) | Volatile reagents with exchangeable deuterons. Enables in situ confirmation of -OH, -NH, -COOH via D 1s signal or O 1s BE shift. | Confirming hydrogen-bonded physisorption vs. covalent chemisorption. |
| Certified Reference Materials | Well-characterized surfaces (e.g., Au foil, SiO2/Si wafer, highly ordered pyrolytic graphite - HOPG). Provides substrate BE and morphology control. | Validating instrument performance and charge referencing protocol. |
| UHV-Compatible Heater Stage | Sample stage capable of controlled heating (ambient to 600°C) under UHV. Tests thermal stability of adsorbed layer. | Distinguishing physisorbed (desorbs at low T) from chemisorbed species. |
| Low-Energy Electron Flood Gun | Source of low-energy (0.1-10 eV) electrons to neutralize charge on insulating samples. Critical for accurate BE on drug-polymer composites. | Analyzing adsorption on polymeric drug delivery carriers. |
| Gas Dosing System | UHV manifold with leak valve for controlled exposure to reactive (O2, H2) or inert (Ar) gases. | Studying in situ oxidation or reduction of chemisorbed catalytic layers. |
Case Study: Drug on Polymer Surface
Scenario: Distinguishing chemisorbed (amide-linked) from physisorbed (hydrogen-bonded) antibody on a polymer-coated medical device.
- ARXPS: The N 1s (amide) to polymer C 1s ratio remains constant at 0°, 45°, and 70°. This suggests the antibody is uniformly distributed within the analysis depth, consistent with a thin, conformal chemisorbed layer.
- In Situ Treatment: Gentle heating to 120°C causes no change in N 1s intensity or shape, ruling out physisorption.
- Peak Deconvolution: High-resolution N 1s is fit with two components: amide (-NH-CO-) at 399.9 eV and amine/protonated amine at 399.1/401.2 eV. The relative areas match the expected antibody structure, validating the assignment.
Beyond XPS Alone: Validating Adsorption Mechanisms with Complementary Analytical Techniques
Correlating XPS Data with ToF-SIMS for Molecular Fragment Identification
Within the broader thesis on X-ray Photoelectron Spectroscopy (XPS) analysis for differentiating and characterizing physisorbed and chemisorbed species on material surfaces, this application note addresses a central challenge: the unambiguous identification of molecular fragments. While XPS provides superb quantitative elemental and chemical state information, its ability to identify specific molecular ions or complex organic fragments is limited. Time-of-Flight Secondary Ion Mass Spectrometry (ToF-SIMS) excels at detecting molecular and fragment ions with high mass resolution and sensitivity. Correlating data from these two techniques is therefore critical for constructing a complete molecular picture of adsorbed species, distinguishing between weakly bound physisorbed layers and strongly bound chemisorbed monolayers, which is fundamental in drug delivery system development, biomaterial interface studies, and catalyst research.
Core Principles of Correlation
Table 1: Complementary Strengths of XPS and ToF-SIMS
| Technique | Analytical Information | Depth Resolution | Quantification | Molecular Specificity | Damage Risk |
|---|---|---|---|---|---|
| XPS | Elemental composition, oxidation states, chemical bonding. | 5-10 nm (with angle-resolved). | Excellent (atomic %). | Low (functional groups only). | Low (X-ray induced damage possible for organics). |
| ToF-SIMS | Elemental & molecular ion masses, fragment patterns, mapping. | 1-3 nm (static mode). | Semi-quantitative (matrix effects). | Excellent (up to ~1000 m/z). | High (ion beam can degrade organics). |
The correlation strategy involves using XPS to define the elemental stoichiometry and gross chemical environment (e.g., C-C/C-H vs C-O vs C=O, metal oxidation state). This quantitative framework then constrains the interpretation of the complex ToF-SIMS fragment patterns, allowing researchers to pinpoint which molecular fragments are consistent with the chemical states identified by XPS and to assign them to either the adsorbed species or the substrate.
Experimental Protocol for Correlated Analysis
Protocol 1: Sequential Analysis of Adsorbed Molecular Layers Objective: To characterize the composition, bonding, and fragmentation behavior of a self-assembled monolayer (SAM) on a gold substrate, serving as a model for a chemisorbed system.
Materials & Pre-Analysis:
- Sample: Gold-coated silicon wafer functionalized with a candidate drug-like thiolate SAM.
- Sample Handling: Use clean, powder-free gloves and tweezers. Store and transfer in a dedicated nitrogen-purged container.
- Preliminary Step: Record optical or SEM images to define analysis regions of interest (ROIs).
Step-by-Step Procedure:
- Introduction into UHV:
- Load the sample into the introduction chamber of the integrated XPS-ToF-SIMS system or a dedicated transfer vessel.
- Pump down to a base pressure ≤ 1 x 10⁻⁷ mbar.
- Transfer to the analysis chamber (pressure ≤ 5 x 10⁻⁹ mbar).
XPS Analysis (Perform First - Less Damaging):
- Survey Spectrum: Acquire over a binding energy range of 0-1100 eV with pass energy of 100-150 eV. Identify all elements present.
- High-Resolution Spectra: Acquire for Au 4f, S 2p, C 1s, N 1s, O 1s (or other relevant elements) with pass energy of 20-50 eV for optimal resolution.
- Data Processing: Apply charge correction (e.g., reference Au 4f₇/₂ to 84.0 eV). Fit high-resolution spectra using appropriate background (Shirley or Tougaard) and Gaussian-Lorentzian line shapes.
- Output: Quantified atomic percentages and chemical state assignments (e.g., S 2p doublet position confirms thiolate (chemisorbed) vs. physisorbed sulfur species).
ToF-SIMS Analysis (Perform in Same ROI):
- Instrument Setup: Use a pulsed Bi₃⁺ or Gas Cluster Ion Beam (GCIB) primary ion source for reduced fragmentation of organics. Ensure the primary ion dose remains in the "static SIMS" regime (< 10¹² ions/cm²).
- Spectral Acquisition: Acquire positive and negative ion spectra from the identical ROI analyzed by XPS. Use high mass resolution mode (m/Δm > 5000).
- Data Acquisition: Collect spectra for adequate primary ion dose to ensure good signal-to-noise for molecular ions.
- Data Processing: Calibrate mass scale using known peaks (e.g., CH₃⁺, C₂H₅⁺, Au⁺). Identify molecular ions and fragments (e.g., [M+H]⁺, [M-H]⁻, characteristic fragment clusters).
Correlation & Interpretation:
- Use the S/Au ratio from XPS to estimate SAM coverage.
- Use the C 1s peak component ratios (C-C, C-O, C=O) from XPS to validate the relative intensities of related fragments in ToF-SIMS (e.g., CₓHᵧ⁺ vs. CₓHᵧO⁺).
- Identify the molecular ion peak in ToF-SIMS that corresponds to the intact chemisorbed molecule minus the anchoring group (e.g., [M - H]⁻ for a thiol) and correlate its presence/absence with the chemical state of sulfur from XPS.
Protocol 2: Differentiating Physisorbed vs. Chemisorbed Layers via Sputtering Objective: To distinguish between a physisorbed protein layer and a chemisorbed linker molecule on a titanium oxide surface.
Procedure:
- Perform XPS and ToF-SIMS analysis on the "as-received" surface as per Protocol 1.
- Subject the sample to a gentle, low-energy Ar⁺ sputter (e.g., 500 eV, 1-2 minutes) in the analysis chamber.
- Repeat XPS and ToF-SIMS analysis on the sputtered region.
- Correlation: A chemisorbed species (e.g., phosphonate linker) will show persistent XPS signals (Ti-O-P, P 2p) and characteristic ToF-SIMS fragments (e.g., PO₂⁻, PO₃⁻) after sputtering. A physisorbed layer (e.g., adsorbed BSA protein) will show a dramatic decrease in XPS N 1s signal and the disappearance of high-mass molecular ions/cluster ions (e.g., amino acid fragments) in ToF-SIMS.
Diagram 1: Workflow for correlating XPS and ToF-SIMS data.
The Scientist's Toolkit: Key Research Reagent Solutions
Table 2: Essential Materials for XPS-ToF-SIMS Correlation Studies
| Item | Function & Relevance |
|---|---|
| Certified Reference Materials (e.g., Au, Si, Highly Ordered Pyrolytic Graphite) | For daily instrument performance checks, energy scale calibration (XPS), and mass scale calibration (ToF-SIMS). Essential for reproducible, comparable data. |
| Model Substrates (e.g., Atomically flat Si wafers, template-stripped Au, single crystal TiO₂) | Provide ultra-clean, well-defined surfaces for controlled adsorption studies, minimizing spectral interference from substrate roughness or contamination. |
| Well-Characterized Adsorbates (e.g., Alkanethiols, Silanes, Peptides with known sequences) | Act as calibration standards for chemisorption. Their known structure provides reference XPS spectra and characteristic ToF-SIMS fragmentation patterns. |
| Charge Neutralization Systems (Flood guns, low-energy electron/ion beams) | Critical for analyzing insulating samples (e.g., polymers, biological layers) with both techniques to prevent peak shifting (XPS) and distortion (ToF-SIMS). |
| Gas Cluster Ion Beam (GCIB) Source | A ToF-SIMS primary ion source that significantly reduces fragmentation of large organic molecules and biomolecules, enabling clearer molecular ion detection for correlation with XPS functional group data. |
| In-Situ Sputter Ion Source | Integrated Ar⁺ or C₆₀⁺ source for controlled depth profiling. Key for the protocol to differentiate physisorbed vs. chemisorbed layers. |
| UHV-Compatible Transfer Vessels | Allow safe, contamination-free transfer of air-sensitive samples (e.g., from solution adsorption cell) or between separate XPS and ToF-SIMS instruments. |
Case Study & Data Correlation Table
Study: Characterization of a Polyethylene Glycol (PEG) silane film on silicon oxide (a common non-fouling coating in drug delivery devices). Question: Is the film chemically grafted (chemisorbed) or physically adsorbed?
Table 3: Correlated Data from PEG Silane on SiO₂
| Analytical Target | XPS Result | ToF-SIMS Result (Positive Mode) | Correlation & Interpretation |
|---|---|---|---|
| Evidence of Grafting | Si 2p shows component at ~102 eV (Si-O-Si from siloxane bond). Atomic % Si decreases after gentle wash. | Intense peaks at m/z 147, 207, 221 (characteristic of SiₓOᵧC₂Hₓ⁺ fragments from the silane headgroup). | Persistent Si-C/O related fragments in SIMS after washing confirm chemisorbed siloxane network. |
| PEG Integrity | C 1s spectrum dominated by C-O component (~286.5 eV). O/C atomic ratio ~0.66 (matches PEG stoichiometry). | Repeat unit clusters: C₂H₅O⁺ (45), C₄H₉O₂⁺ (89), [M+Na]⁺ for full chain. | XPS O/C ratio validates PEG-like ToF-SIMS pattern. High-mass ions confirm intact polymer chains. |
| Contamination (Physisorbed) | Small C 1s component at 285.0 eV (C-C/C-H). | Hydrocarbon series (CₙH₂ₙ₊₁⁺) dominate if present. | Correlation shows if C-C XPS signal correlates with hydrocarbon fragments (physisorbed contaminants) or is part of the PEG backbone (C-O in SIMS). |
Diagram 2: Decision logic for adsorbed layer characterization.
Best Practices and Concluding Remarks
- Order of Analysis: Always perform XPS first on a pristine area, as X-ray beams are typically less damaging than ion beams used in ToF-SIMS.
- Spatial Correlation: Use internal landmarks or a micro-focused XPS source to ensure the exact same region is analyzed by both techniques.
- Data Validation: Use the quantified elemental ratios from XPS to sanity-check the relative peak intensities in ToF-SIMS, acknowledging that SIMS sensitivities vary greatly.
- Conclusion for Thesis Context: The systematic correlation of XPS and ToF-SIMS data provides a powerful, bi-modal verification system. It moves surface analysis beyond simple identification toward definitive molecular assembly understanding, crucially differentiating between transient physisorbed species and stable chemisorbed interfaces—a cornerstone for rational design in drug delivery and biomaterials science.
Using AFM and Ellipsometry to Measure Adsorbate Layer Thickness and Morphology
Within the framework of a thesis investigating X-ray Photoelectron Spectroscopy (XPS) for the characterization of physisorbed and chemisorbed species, Atomic Force Microscopy (AFM) and Spectroscopic Ellipsometry (SE) serve as critical complementary techniques. While XPS excels at providing chemical state and elemental composition data from the top 1-10 nm of a surface, it offers limited direct information on the physical thickness and nanoscale morphology of adsorbed layers. AFM provides three-dimensional topological maps with sub-nanometer vertical resolution, enabling the visualization of adsorbate islands, surface coverage, and roughness. Spectroscopic Ellipsometry provides a rapid, non-contact method for determining the average thickness and optical properties of thin films (from sub-nm to several microns) over a larger sample area. The integration of data from these three techniques (XPS, AFM, SE) yields a comprehensive picture of adsorbate presence, chemistry, thickness, and structure, which is indispensable for research in surface science, materials engineering, and drug development—particularly in studies of protein adsorption, polymer brush layers, self-assembled monolayers (SAMs), and lipid bilayers.
Core Principles and Data Correlation
AFM measures the force between a sharp tip and the surface. In tapping mode, it gently probes the topography, revealing the morphology of adsorbates. Cross-sectional analysis of height profiles directly yields local thickness measurements for isolated features (e.g., nanoparticles, molecular islands).
Spectroscopic Ellipsometry measures the change in polarization state of light reflected from a sample. The measured parameters Psi (Ψ) and Delta (Δ) are modeled to extract the optical constants (refractive index n, extinction coefficient k) and the thickness of thin films. For ultra-thin adsorbate layers, effective medium approximation models are often used.
Correlation with XPS: A measured increase in film thickness via SE or AFM should correlate with an increased signal from adsorbate-specific elements (e.g., N, P for biomolecules) and a attenuated signal from the substrate in XPS. AFM morphology can explain heterogeneous XPS signals.
Table 1: Comparison of AFM and Ellipsometry for Adsorbate Characterization
| Parameter | Atomic Force Microscopy (AFM) | Spectroscopic Ellipsometry (SE) |
|---|---|---|
| Primary Output | 3D Topography map, Roughness (Rq, Ra) | Thickness (d), Refractive index (n), Extinction coeff. (k) |
| Thickness Range | ~0.1 nm to > 10 µm | ~0.1 nm to > 10 µm (depends on model) |
| Lateral Resolution | ~1 nm (ideal) | ~1 mm to µm (spot size, no imaging) |
| Measurement Type | Direct, local | Indirect, area-averaged |
| Sample Environment | Air, liquid, vacuum | Primarily air, liquid cells possible |
| Throughput | Slow (min-hr per scan) | Very Fast (seconds per point) |
| Key Strengths | Direct visualization, nanoscale morphology, operates in fluid | Non-contact, highly precise for uniform films, fast kinetics |
| Key Limitations | Tip convolution effects, slow scanning, can disturb soft layers | Requires optical model, assumes uniformity, lower lateral resolution |
Table 2: Typical Data from Combined Study of a Chemisorbed SAM on Gold
| Analysis Technique | Measured Parameter | Typical Result for C11 SAM | Information Gained |
|---|---|---|---|
| XPS | C 1s / Au 4f ratio | Increased vs. bare Au | Confirms organic layer presence & approximate coverage |
| XPS | S 2p peak position | 162.0 eV (bound thiolate) | Confirms chemisorption via S-Au bond |
| Ellipsometry | Average Thickness | 1.4 ± 0.1 nm | Consistent with all-trans alkyl chain length |
| AFM | RMS Roughness (Rq) | 0.3 nm (vs. 0.2 nm for bare Au) | SAM is conformal, does not increase roughness significantly |
| AFM | Island Height (if incomplete) | 1.5 ± 0.2 nm | Direct local thickness of adsorbate domains |
Experimental Protocols
Protocol 1: Ellipsometry for In-Situ Adsorption Kinetics and Thickness
Objective: To measure the average thickness and adsorption kinetics of a protein layer from solution in real-time.
Materials & Setup:
- Spectroscopic Ellipsometer with liquid cell.
- Polarizer, compensator, sample, analyzer (PCSA) setup.
- Stable temperature controller (±0.1°C).
- Buffer solution (e.g., PBS, pH 7.4).
- Protein solution (e.g., 1 mg/mL Bovine Serum Albumin in PBS).
- Clean, well-characterized substrate (e.g., silicon wafer with oxide layer).
Procedure:
- Substrate Preparation & Baseline: Clean substrate (e.g., piranha etch for Si, UV-Ozone for Au). Mount in liquid cell. Fill cell with pure buffer. Acquire a spectroscopic (e.g., 400-800 nm) ellipsometry measurement (Ψ, Δ) of the substrate/buffer interface. This is the baseline.
- Optical Model Construction: Using modeling software, build a layered optical model: Substrate (Si) / Native Oxide (SiO₂, fixed n,k,d) / Adsorbate Layer (to fit) / Buffer (fixed n,k). Use Cauchy or effective medium approximation for the adsorbate layer initially.
- In-Situ Kinetics Measurement: Set ellipsometer to kinetic mode (single wavelength, e.g., 532 nm). Initiate flow or injection of protein solution into the cell to replace buffer.
- Data Acquisition: Continuously measure Ψ and Δ at 1-5 second intervals for the duration of adsorption (typically 30-60 min).
- Data Analysis: For each time point, fit the model to the measured (Ψ, Δ), allowing the adsorbate layer thickness (d) and sometimes refractive index (n) to vary. Plot d vs. time to obtain the adsorption kinetic curve.
- Post-Adsorption Characterization: Replace with pure buffer to remove loosely bound species. Perform a final spectroscopic scan. Refit the model for accurate final thickness and n.
Protocol 2: AFM for Ex-Situ Morphology and Local Thickness
Objective: To characterize the surface morphology and measure the local thickness of an adsorbed polymer film after drying.
Materials & Setup:
- Atomic Force Microscope (Tapping Mode recommended).
- Sharp silicon cantilevers (resonant frequency 200-400 kHz in air).
- Sample substrates with adsorbed layer.
- Vibration isolation table.
Procedure:
- Sample Preparation: Adsorb the target species onto a flat substrate (e.g., mica for biomolecules, Si wafer for polymers). Rinse gently with appropriate solvent (e.g., deionized water) to remove salts or unbound material. Dry under a gentle stream of inert gas (N₂ or Ar). Note: Drying may alter morphology of soft layers.
- AFM Calibration: Calibrate the AFM scanner using a grating with known step height (e.g., 20 nm, 100 nm).
- Imaging Parameters: Engage the cantilever in tapping mode. Optimize setpoint and drive amplitude to achieve stable, low-force imaging. Select scan areas (e.g., 1 µm x 1 µm, 5 µm x 5 µm, 10 µm x 10 µm).
- Topography Acquisition: Acquire height and phase images at moderate resolution (512 x 512 pixels). Perform multiple scans on different sample areas to ensure reproducibility.
- Scratch Test for Local Thickness (if needed): On a representative area, use the AFM tip in contact mode with high force to scratch through the adsorbate layer. Image a larger area encompassing the scratch.
- Data Analysis:
- Roughness: Use AFM software to calculate the root-mean-square (Rq) and average (Ra) roughness of a selected area.
- Feature Height: Draw line profiles across individual adsorbate features (e.g., islands, particles) to measure their height.
- Scratch Depth: Draw a line profile perpendicular to the scratch. The depth from the top of the film to the exposed substrate is the film thickness. Average multiple measurements.
Visualizations
Title: Complementary Technique Integration for Surface Analysis
Title: Sequential Protocol for Adsorbate Layer Characterization
The Scientist's Toolkit
Table 3: Essential Research Reagent Solutions & Materials
| Item | Function / Role in Experiment |
|---|---|
| Ultra-Flat Substrates (Si wafers, Mica, Template-stripped Au) | Provides an atomically smooth, reproducible baseline for thickness and morphology measurements, minimizing substrate roughness interference. |
| Piranha Solution (3:1 H₂SO₄:H₂O₂) | Caution: Highly hazardous. Used to clean silicon/glass substrates, creating a hydrophilic, contaminant-free surface with active -OH groups. |
| Alkanethiols (e.g., 1-Octadecanethiol, C18) | Model chemisorbing molecules to form Self-Assembled Monolayers (SAMs) on gold for technique calibration and controlled studies. |
| Phosphate Buffered Saline (PBS), pH 7.4 | Standard physiological buffer for biological adsorption studies (e.g., proteins, antibodies), maintaining biomolecule stability. |
| Bovine Serum Albumin (BSA) Solution | A model "sticky" protein used as a standard for testing and calibrating adsorption measurement protocols. |
| Polystyrene Beads (e.g., 100 nm diameter) | Monodisperse particles used as height standards for AFM calibration and as model adsorbates for morphology studies. |
| Ellipsometry Modeling Software (e.g., CompleteEASE, WVASE) | Essential for converting raw (Ψ, Δ) data into physical parameters (thickness, n, k) by fitting to an optical layer model. |
| Sharp AFM Probes (Si, freq. ~300 kHz) | High-resolution tips for tapping mode AFM, essential for accurate imaging of nanoscale adsorbate features without excessive tip convolution. |
Employing Contact Angle Measurements to Correlate XPS Composition with Surface Wettability
Within a broader thesis investigating X-ray Photoelectron Spectroscopy (XPS) analysis for physisorbed and chemisorbed species, establishing a quantitative link between surface chemical composition and macroscopic wettability is paramount. Contact angle (CA) measurement provides a direct, rapid assessment of surface wettability, which is governed by the outermost atomic layers (typically <10 nm) probed by XPS. Correlating XPS-derived atomic percentages and functional group identification with CA data enables researchers to deconvolute the contributions of chemisorbed monolayers from physisorbed contaminants, and to engineer surfaces with tailored wettability for applications in drug delivery, implant biocompatibility, and diagnostic device development.
Core Principles and Data Correlation
Surface wettability, quantified by the water contact angle (θ), is described by models such as Young’s equation for ideal solids and Wenzel or Cassie-Baxter models for rough surfaces. The critical surface tension (γ_c) derived from Zisman plots further characterizes wettability. XPS determines the elemental and chemical state composition of this same surface region. A direct correlation exists between the concentration of non-polar groups (e.g., C-C/C-H from hydrocarbon contamination or intentional coatings) and increased hydrophobicity (high θ), while polar groups (e.g., -OH, -COOH, -NH2) enhance hydrophilicity (low θ).
Table 1: Correlation of XPS-Derived Functional Groups with Wettability Trends
| XPS-Detected Functional Group / Elemental Ratio | Typical Binding Energy (eV) | Effect on Water Contact Angle | Common Origin in Surface Science |
|---|---|---|---|
| C-C / C-H (hydrocarbon) | C 1s: 284.8 | Strong Increase (Hydrophobic) | Adventitious carbon, alkyl SAMs, polymer backbones |
| -CF2- / -CF3 | C 1s: ~291.0 (CF2), ~293-294 (CF3); F 1s: ~689 | Very Strong Increase (Superhydrophobic) | Fluorinated coatings (e.g., Teflon-like films) |
| C-O / C-OH | C 1s: 286.1-286.5 | Moderate Decrease (Hydrophilic) | Alcohols, contaminants, PEG chains |
| C=O / O-C=O | C 1s: 287.8-289.0 | Decrease (Hydrophilic) | Aldehydes, ketones, carboxylic acids |
| -COOH | C 1s: 288.9-289.2; O 1s: ~531.3 | Strong Decrease (Hydrophilic) | Carboxylic acid-terminated SAMs, oxidized surfaces |
| -NH2 | N 1s: ~399.3-399.5 | Decrease (Hydrophilic) | Aminated surfaces (e.g., for protein coupling) |
| SiO2 (highly networked) | Si 2p: 103.3-103.5 | Decrease (Hydrophilic) | Clean glass, plasma-oxidized silicon |
| O/C Atomic Ratio (from XPS) | N/A | Higher Ratio → Lower θ | Indicator of overall surface polarity |
Detailed Experimental Protocols
Protocol 3.1: Integrated Sample Preparation for XPS and CA Analysis
Objective: To prepare identical, well-defined surfaces for sequential, correlative analysis.
- Substrate Cleaning: Clean substrate (e.g., Si wafer, glass slide, polymer film) via sequential sonication in acetone, isopropanol (10 min each), and rinse with ultrapure water (18.2 MΩ·cm). Dry under a stream of filtered N2 or Ar.
- Surface Modification: Apply the desired modification (e.g., oxygen plasma treatment for 30-120 s, silanization, polymer spin-coating, protein adsorption) to a batch of identical samples.
- Sample Division: Divide the batch into matched pairs. One set is for XPS, the other for CA. Crucially, both sets must be prepared simultaneously under identical conditions.
- Storage and Transfer: Store samples in a clean, dry environment. Transfer to analysis instruments in a controlled manner (e.g., in a vacuum desiccator or under inert atmosphere) to minimize adventitious adsorption prior to measurement. Measure CA first on its set, then analyze the matching set by XPS to avoid X-ray-induced surface damage affecting wettability.
Protocol 3.2: Static Sessile Drop Contact Angle Measurement
Objective: To quantify the equilibrium wettability of the prepared surface.
- Instrument Setup: Level the stage of a goniometer. Set environmental conditions (temperature, humidity) and record them.
- Sample Mounting: Secure the sample flat on the stage. For powders, press into a pellet.
- Dispensing: Using a micro-syringe, dispense a 2-5 µL ultrapure water droplet onto the surface. Ensure the needle tip is close to the surface before droplet release.
- Image Capture: Capture a high-contrast side-view image within 3 seconds of droplet deposition to minimize evaporation effects.
- Angle Analysis: Use instrument software to fit the droplet profile (Young-Laplace or tangent method) and determine the left and right contact angles. Report the average of at least 5 measurements on different spots per sample.
Protocol 3.3: XPS Analysis for Surface Composition
Objective: To determine the elemental and chemical state composition of the top 5-10 nm of the sample.
- Sample Loading: Load the CA-matched sample into the XPS introduction chamber without touching the analysis area. Begin pump-down.
- Survey Scan: Acquire a wide energy survey scan (e.g., 0-1100 eV, pass energy 150 eV) to identify all elements present.
- High-Resolution Scans: Acquire high-resolution spectra (pass energy 20-50 eV) for relevant core levels (C 1s, O 1s, N 1s, etc.). Use sufficient scans for good signal-to-noise.
- Charge Neutralization: Use low-energy electron/ion flood guns for insulating samples to maintain consistent energy calibration. Reference adventitious C 1s to 284.8 eV.
- Quantification & Fitting: Calculate atomic percentages using instrumental sensitivity factors. Deconvolute high-resolution spectra (e.g., C 1s) into sub-peaks representing different functional groups using appropriate software (e.g., CasaXPS, Avantage). Constrain fits with realistic full-width half-maximum and chemically logical binding energies.
Protocol 3.4: Data Correlation and Zisman Plot Construction
Objective: To empirically relate surface composition to critical surface tension.
- Create a Series: Use a set of surfaces with graded composition (e.g., plasma-treated polymers with varying degradation times, mixed SAMs).
- Measure Cos θ vs. γLV: Measure CA (θ) for a series of liquids with known, varying surface tensions (γLV) (e.g., water, diiodomethane, formamide, ethylene glycol).
- Plot & Linear Fit: For each surface, plot cos θ against γ_LV. Perform a linear fit.
- Determine γc: Extrapolate the fit to cos θ = 1. The corresponding γLV value is the critical surface tension (γ_c) of the solid.
- Correlate with XPS: Plot γ_c or cos θ of water against XPS-derived parameters (e.g., O/C atomic ratio, % area of C-O component) for the surface series. Perform linear or non-linear regression to establish the correlation.
The Scientist's Toolkit: Research Reagent Solutions
Table 2: Essential Materials for XPS-CA Correlation Studies
| Item | Function / Purpose |
|---|---|
| Ultrapure Water (Type I, 18.2 MΩ·cm) | Standard liquid for CA; ensures minimal ionic contamination affecting surface tension. |
| High-Purity Solvents (HPLC Grade Acetone, IPA) | Substrate cleaning to remove organic contaminants prior to surface modification. |
| Reference Liquids for Surface Energy (Diiodomethane, Ethylene Glycol, Formamide) | Used in multi-liquid CA methods to calculate surface energy components and construct Zisman plots. |
| Silane Coupling Agents (e.g., Octadecyltrichlorosilane, (3-Aminopropyl)triethoxysilane) | To create well-defined, chemisorbed monolayers with specific terminal functionalities (CH3, NH2) for model studies. |
| Plasma Cleaner (Oxygen or Air Plasma) | Generates a highly reproducible, clean, and hydrophilic surface by introducing polar oxygen-containing groups. |
| Standard Reference Samples (Clean Si Wafer, PTFE Sheet) | Provide known, stable contact angles (Si/SiO2: ~0-10°, PTFE: ~108-112°) for goniometer calibration and validation. |
| Conductive Adhesive Tape (Carbon or Copper) | For mounting insulating samples in XPS to minimize charging, ensuring reliable spectral data. |
| Charge Neutralization Standards (e.g., Freshly Cleaned Gold Foil) | Used to verify and calibrate the charge neutralization system on the XPS instrument. |
Workflow for Correlating CA and XPS Data (96 chars)
Logic of Surface Property Prediction (84 chars)
Comparing XPS Results Before and After Controlled Solvent Washing to Test Adsorption Strength
This application note details a critical protocol for distinguishing physisorbed from chemisorbed species in surface science research, a central theme in a broader thesis utilizing X-ray Photoelectron Spectroscopy (XPS). Controlled solvent washing acts as a selective, non-destructive probe for adsorption strength. Physisorbed species, bound by weak van der Waals forces, are typically removed by washing with an appropriate solvent, while chemisorbed species, involving stronger covalent or ionic bonds, remain intact. The comparative XPS analysis before and after washing provides quantitative, surface-specific evidence of adsorption mechanism and strength, which is vital for applications in drug delivery (protein corona studies), catalyst development, and biomaterial surface modification.
Experimental Protocols
Protocol 1: Sample Preparation & Adsorption
Objective: To create a surface with a mixture of adsorbed species for testing.
- Substrate Cleaning: Clean the substrate (e.g., gold-coated slide, silicon wafer) via successive sonication in acetone, isopropanol, and deionized water (5 minutes each). Dry under a stream of inert gas (N₂ or Ar).
- Adsorption Solution Preparation: Prepare a solution containing the analyte of interest (e.g., a target drug molecule, protein, or polymer) in a suitable solvent at a known concentration (e.g., 1 mg/mL in PBS or ethanol).
- Incubation: Immerse the clean substrate in the adsorption solution for a controlled period (e.g., 1-24 hours) at a defined temperature (e.g., 25°C).
- Initial Rinse: Gently remove the sample and rinse the surface with pure solvent (3x) to remove loosely associated bulk solution. Gently dry under inert gas. This is the "Before Wash" sample.
Protocol 2: Controlled Solvent Washing
Objective: To selectively desorb physisorbed species without altering the substrate or chemisorbed layer.
- Solvent Selection: Choose a solvent that is a good dispersant for the analyte but does not chemically react with the substrate or chemisorbed monolayer. Common choices include ethanol, methanol, toluene, or PBS, depending on system hydrophilicity/hydrophobicity.
- Washing Procedure: Place the "Before Wash" sample in a vial. Add 10-20 mL of the chosen wash solvent.
- Agitation: Subject the sample to gentle agitation for a set duration (e.g., 10 minutes on an orbital shaker at 100 rpm) or sequential static immersions with fresh solvent.
- Drying: Carefully remove the sample, rinse once with fresh solvent to displace any residual, and dry thoroughly under a stream of inert gas. This is the "After Wash" sample.
Protocol 3: XPS Analysis & Data Comparison
Objective: To quantitatively compare surface composition and chemical states.
- Instrument Setup: Load "Before Wash" and "After Wash" samples into the XPS instrument. Use a monochromatic Al Kα X-ray source (1486.6 eV).
- Survey Spectra Acquisition: Acquire wide survey spectra (e.g., 0-1100 eV binding energy) at a pass energy of 160 eV to identify all elements present.
- High-Resolution Spectra Acquisition: For elements of interest (e.g., C 1s, N 1s, O 1s, specific metal peaks), acquire high-resolution spectra at a pass energy of 20-40 eV.
- Data Processing: Apply consistent charge correction (e.g., referencing adventitious C 1s to 284.8 eV). Fit high-resolution peaks using appropriate software (e.g., CasaXPS, Avantage).
- Quantification: Calculate atomic concentrations (%) from peak areas using relative sensitivity factors (RSFs).
Data Presentation
Table 1: Comparative Atomic Concentration (%) from XPS Survey Scans
| Element | Before Wash (At. %) | After Wash (At. %) | Δ (At. %) | % Change |
|---|---|---|---|---|
| C 1s | 65.2 | 58.1 | -7.1 | -10.9% |
| N 1s | 12.5 | 10.8 | -1.7 | -13.6% |
| O 1s | 20.1 | 18.5 | -1.6 | -8.0% |
| Au 4f | 2.2 | 12.6 | +10.4 | +472.7% |
Table 2: High-Resolution C 1s Peak Component Analysis
| Component (Binding Energy) | Assignment | Before Wash (% of C 1s) | After Wash (% of C 1s) | Notes |
|---|---|---|---|---|
| C-C/C-H (284.8 eV) | Aliphatic | 45% | 55% | |
| C-O/C-N (286.3 eV) | Alcohol, Amine | 30% | 25% | Decrease suggests removal of physisorbed species. |
| C=O (288.0 eV) | Amide, Carboxyl | 20% | 17% | |
| O-C=O (289.2 eV) | Ester, Acid | 5% | 3% |
Mandatory Visualizations
Title: XPS Adsorption Strength Testing Workflow
Title: Interpreting XPS Signal Changes After Washing
The Scientist's Toolkit
Table 3: Essential Research Reagents & Materials
| Item | Function in Protocol |
|---|---|
| XPS Instrument (with Al Kα source) | Provides core-level spectra to identify elements and chemical states on the top 1-10 nm of the surface. |
| Ultra-pure Solvents (Acetone, IPA, Toluene, Ethanol) | For substrate cleaning and selective desorption of physisorbed species. Must be high purity to avoid contamination. |
| Inert Gas Drying Gun (N₂ or Ar) | Provides a clean, moisture-free stream for drying samples without leaving residues or oxidizing surfaces. |
| Analytical Balance (0.01 mg sensitivity) | Accurate preparation of adsorption solutions at precise concentrations. |
| Ultrasonic Cleaner | For thorough and consistent substrate cleaning prior to adsorption experiments. |
| Adventitious Carbon Reference Standard (e.g., cleaned, oxidized Si wafer) | Aids in consistent charge correction of XPS spectra by providing a known C 1s peak position (284.8 eV). |
| Peak Fitting Software (e.g., CasaXPS) | Enables deconvolution of complex high-resolution XPS peaks to quantify different chemical states of an element. |
Within a broader thesis on utilizing X-ray Photoelectron Spectroscopy (XPS) for characterizing physisorbed and chemisorbed species, this document provides critical application notes and protocols. A cohesive model of surface modification for drug delivery carriers (e.g., polymeric nanoparticles, liposomes, mesoporous silica) requires integrating multi-technique data to distinguish between reversible physisorption and stable chemisorption of targeting ligands, stealth polymers, and drug molecules. XPS serves as a cornerstone technique for quantifying elemental surface composition, identifying chemical states, and providing evidence for covalent grafting versus physical adherence.
Application Notes: Key Data & Interpretation
Successful modeling hinges on correlating data from XPS with other surface-sensitive and bulk techniques. The following table summarizes primary quantitative metrics and their significance.
Table 1: Integrated Data Matrix for Surface Modification Analysis
| Analytical Technique | Primary Quantitative Output | Information for Cohesive Model | Distinguishing Physisorption vs. Chemisorption |
|---|---|---|---|
| X-ray Photoelectron Spectroscopy (XPS) | Atomic % (e.g., N, S, P), binding energy shifts (ΔBE in eV), peak area ratios. | Surface elemental composition, chemical bonding states, layer thickness (via angle-resolved). | Covalent bonding shows characteristic BE shifts (e.g., C-N/C-O). Physisorption may show signal from underlying substrate. |
| Time-of-Flight Secondary Ion Mass Spectrometry (ToF-SIMS) | Characteristic fragment ion intensities (counts). | Molecular fingerprint of surface species, imaging of distribution. | Presence of unique fragment ions from linker chemistry confirms chemisorption. |
| Quartz Crystal Microbalance with Dissipation (QCM-D) | Frequency change (ΔF, Hz), Dissipation change (ΔD). | Mass adsorbed (including hydrodynamically coupled solvent), viscoelastic properties. | Rigid, low-ΔD layers suggest chemisorbed monolayers. High-ΔD, thick layers suggest physisorbed, swollen layers. |
| Surface Plasmon Resonance (SPR) | Resonance unit change (ΔRU), kinetic rate constants (ka, kd). | Real-time adsorption mass, binding affinity. | Irreversible binding (very low kd) suggests chemisorption. Reversible binding suggests physisorption. |
| Fluorescence Spectroscopy / Confocal Microscopy | Fluorescence intensity, co-localization coefficients. | Confirmation of ligand binding and cellular uptake in vitro. | Failure of harsh washes to remove signal indicates stable chemisorption. |
Experimental Protocols
Protocol 3.1: XPS Analysis of PEGylated Gold Nanoparticles (Model System)
Objective: To confirm chemisorption of thiol-terminated polyethylene glycol (SH-PEG) onto gold nanoparticle (AuNP) surfaces and quantify surface coverage.
Sample Preparation:
- Synthesis: Synthesize 50 nm citrate-stabilized AuNPs via the Turkevich method.
- Modification: Incubate 1 mL of AuNP solution with 1 mM SH-PEG (5 kDa) for 24 hours at room temperature under gentle agitation.
- Purification: Remove excess ligand by triple centrifugation (14,000 rpm, 20 min) and resuspension in deionized water. Store final conjugate at 4°C.
XPS Measurement:
- Substrate: Deposit 50 µL of purified AuNP@PEG solution onto a clean silicon wafer and air-dry.
- Instrument Setup: Use a K-Alpha+ XPS system (Thermo Scientific) with monochromatic Al Kα X-rays (1486.6 eV).
- Acquisition Parameters: Survey spectrum: pass energy 150 eV, step size 1 eV. High-resolution spectra for Au 4f, S 2p, C 1s, O 1s: pass energy 50 eV, step size 0.1 eV. Charge compensation with flood gun.
- Analysis: Calculate atomic % from survey spectra. Deconvolute S 2p peak: presence of S 2p3/2 at ~162 eV (Au-S bond) confirms chemisorption. The absence of a peak at ~168 eV (oxidized sulfur) indicates successful purification.
Protocol 3.2: Integrated QCM-D & XPS Protocol for Polymer Film Grafting
Objective: To correlate real-time adsorption kinetics with chemical surface state for polydopamine (PDA) coating and subsequent covalent immobilization of an RGD peptide.
QCM-D Experimental Workflow:
- Baseline: Establish stable baseline in 10 mM HEPES buffer, pH 7.4, at 25°C in QCM-D flow chamber.
- PDA Deposition: Flow 0.5 mg/mL dopamine solution in 10 mM Tris buffer, pH 8.5, for 30 min. Monitor ΔF and ΔD.
- Rinse: Switch to HEPES buffer to remove physisorbed species.
- Peptide Grafting: Activate PDA surface by flowing 10 mM EDC/NHS in MES buffer for 15 min. Flow 0.1 mg/mL amine-terminated RGD peptide solution for 60 min.
- Final Rinse: Rinse with buffer and then DI water. Dry under N2 stream.
Post-QCM-D XPS Analysis:
- Extract the sensor crystal from the module.
- Acquire XPS spectra as per Protocol 3.1, focusing on N 1s peak. Deconvolute to show components: amine (~399.5 eV) from PDA and potential amide (~400.2 eV) from peptide covalent bond.
Visualizations
Title: Integrated Surface Analysis Workflow
Title: Data Integration to Model Outputs
The Scientist's Toolkit: Research Reagent Solutions
Table 2: Essential Materials for Surface Modification & Analysis
| Item | Function / Role | Example/Note |
|---|---|---|
| Thiol-Terminated PEG (SH-PEG) | Forms chemisorbed, biocompatible stealth layer on gold surfaces. | Varies by molecular weight (2k-20k Da). Include -OCH3 or -COOH end groups for further conjugation. |
| (3-Aminopropyl)triethoxysilane (APTES) | Coupling agent for introducing amine groups onto oxide surfaces (SiO2, TiO2). | Enables subsequent covalent immobilization via EDC/NHS chemistry. |
| Polydopamine (PDA) Precursor | Versatile, adherent coating for secondary reaction on virtually any material. | Formed by oxidative polymerization of dopamine. Provides universal platform for grafting. |
| EDC & NHS Crosslinkers | Activate carboxyl groups for covalent amide bond formation with amines. | Critical for conjugating biomolecules (peptides, antibodies) to functionalized surfaces. |
| Certified XPS Reference Samples | Calibration of instrument binding energy scale and verification of resolution. | Clean Au foil (Au 4f7/2 = 84.0 eV), clean Cu foil (Cu 2p3/2 = 932.67 eV). |
| Ultrasensitive QCM-D Sensor Crystals (SiO2-coated) | Real-time, label-free measurement of adsorbed mass and film rigidity. | Typically gold-coated quartz crystals with a silica surface layer. |
Conclusion
Accurate XPS analysis of physisorbed and chemisorbed species is fundamental for advancing biomaterial and drug development. By understanding the foundational spectral signatures, implementing rigorous methodologies, and proactively troubleshooting artifacts, researchers can confidently interpret surface composition. Crucially, validating XPS findings with complementary techniques provides a robust, multi-modal understanding of surface interactions. This integrated approach is essential for designing next-generation implants with controlled bio-interfaces, optimizing drug loading and release from carrier systems, and developing sensitive diagnostic surfaces. Future directions point toward in-situ/operando XPS for dynamic adsorption studies and leveraging advanced machine learning tools for rapid, high-fidelity spectral analysis of complex biological interfaces.